







Geographic atrophy (GA), the advanced stage of dry age-related macular degeneration (AMD), causes progressive and irreversible vision loss.1 Unlike wet AMD, which has benefited from effective anti-VEGF therapies, GA remained largely untreatable until recently. The emergence of complement inhibitors has provided modest benefits, but these single-pathway approaches have significant limitations: continued disease progression, high treatment burden requiring frequent injections, and increased risk of neovascular conversion. Importantly, the European Medicines Agency rejected both approved complement inhibitors’ marketing applications, citing unfavorable benefit-risk profiles due to lack of functional visual benefit.
A novel therapeutic strategy combining neuroprotection and complement modulation through dual-pathway gene therapy represents a paradigm shift in GA treatment. This approach, being developed by Ikarovec Limited, addresses multiple pathological mechanisms simultaneously with a single administration, potentially offering both improved efficacy and reduced treatment burden.
Understanding GA Pathophysiology
GA pathogenesis involves intricate interactions between oxidative stress, inflammation, complement activation, and cellular death pathways. The disease begins with metabolic stress in the retinal pigment epithelium (RPE). High metabolic demands from phototransduction, combined with environmental stressors like smoking and chronic light exposure, lead to accumulation of cellular debris.
Two key pathologic deposits characterize GA progression. Drusen accumulate between the RPE and Bruch’s membrane, consisting of cellular debris, lipids, lipoproteins, and amyloid deposits. Lipofuscin, containing byproducts of photoreceptor outer segment degradation including A2E, builds up within RPE cells. By age 90, lipofuscin granules occupy approximately 20% of macular RPE cell area.
These deposits trigger inflammatory cascades through multiple pathways, including the NLRP3 inflammasome and complement system.2,3 When regulatory mechanisms become compromised (particularly with genetic risk factors affecting complement regulation) chronic inflammation leads to characteristic retinal cell death. The disease’s insidious progression results in expanding atrophic lesions that ultimately involve the fovea, causing severe central vision loss.

Figure 1. CD46 (membrane cofactor protein, MCP) is a ubiquitously expressed cell-surface complement regulator that limits complement activation at the level of C3 and C4. As shown, CD46 acts as an essential cofactor for factor I (FI), enabling the proteolytic inactivation of C3b to iC3b (and downstream C3 fragments) and C4b to inactive products on host cell membranes. By promoting irreversible cleavage of surface-bound C3b, CD46 suppresses formation and stability of both alternative pathway C3/C5 convertases (C3bBb, C3bBbC3b) and classical/lectin pathway convertases (C4b2a, C4b2aC3b), thereby constraining the amplification loop and downstream generation of C5a and membrane attack complex (MAC). Adapted from Harris et al, Molecular Immunology 102 (2018) 89–119. Licensed for reuse via a Creative Commons Attribution License (CC BY).
Complement System Dysregulation in GA
The complement system, consisting of more than 30 proteins involved in pathogen detection and removal, plays a central role in GA pathogenesis.4 Three distinct activation pathways (classical, lectin, and alternative) converge at complement factor C3 cleavage, producing inflammatory C3a and opsonizing C3b fragments. The alternative pathway, being the evolutionarily oldest and undergoing continuous low-level activation, serves as both an independent pathway and an amplification loop for the others.
In healthy eyes, endogenous regulators prevent complement-mediated damage to host cells. Membrane cofactor protein (MCP/CD46) serves as a crucial regulator, acting as a cofactor for complement factor I (CFI) to cleave and inactivate C3b and C4b deposits on cell surfaces.5,6 This rapid inactivation localizes complement activation to pathogens while protecting host tissues.
However, in GA, this protective mechanism fails. RPE cells demonstrate significantly reduced CD46 expression at both mRNA and protein levels very early in disease development, making them vulnerable to complement-mediated damage.7-9 This reduction appears to be both a cause and consequence of GA progression, creating a pathologic cycle where decreased protection leads to increased complement activation and further cellular damage. Notably, reduced expression of CD46 by the RPE/choroid results in drusen-type pathology in mice,10 whereas CD46 expression protects against laser-induced choroidal neovascularization in experimental models.11,12
The Neuroprotective Role of PEDF
Pigment epithelium–derived factor (PEDF) is significantly depleted early in GA development and represents another critical component in GA pathophysiology.13,14 This 50 kDa multifunctional protein maintains RPE and photoreceptor homeostasis through several mechanisms.15 PEDF receptors exist at high density in the RPE cell layer, suggesting important autocrine and paracrine neuroprotective functions in retinal tissue.
PEDF’s neuroprotective properties are particularly relevant to GA. The protein protects human RPE cells against oxidative stress resulting from lipofuscin accumulation, reduces apoptosis, decreases proinflammatory cytokine expression, and helps prevent lipofuscin buildup in experimental models.16-18 PEDF also protects photoreceptors; in animal models, lack of PEDF leads to photoreceptor degeneration,19 while restoration of PEDF protects photoreceptors in murine and human models of retinal degeneration.20 These functions directly address key pathological mechanisms in GA development and progression.
Additionally, PEDF possesses anti-angiogenic properties that may help prevent conversion to neovascular AMD. The protein’s deficiency in patients with neovascular AMD suggests that lack of PEDF activity creates a permissive environment for choroidal neovascularization.21 This is particularly significant given that GA patients face approximately 9% annual risk of developing neovascular AMD.22 The protein’s extremely short half-life of approximately 35 minutes makes it unsuitable for recombinant protein therapy, necessitating gene therapy approaches for sustained therapeutic levels.15

Figure 2. Dual-pathway therapeutic mechanism of combined CD46 and PEDF gene therapy for geographic atrophy (GA). GA pathophysiology involves complement overactivation through the C3-C5-MAC cascade, leading to chronic inflammation, increased angiogenic drive, and photoreceptor degeneration (A). Soluble CD46 (membrane cofactor protein) acts as a cofactor for complement factor I, catalytically inactivating C3b and C4b to suppress complement activation and MAC formation, thereby reducing inflammation (B1). PEDF provides direct neuroprotection, promoting neuronal survival while inhibiting VEGF-mediated angiogenesis (B2). The dual-pathway gene therapy approach (center) combines both mechanisms, simultaneously decreasing complement activation and MAC deposition, enhancing neuroprotection and cellular survival, and reducing both inflammation and angiogenic conversion risk.
Current Treatment Limitations
The FDA has approved 2 complement inhibitors for GA treatment, both representing significant advances but with notable limitations. Pegcetacoplan (Syfovre; Apellis Pharmaceuticals), a C3 inhibitor administered every 4 to 8 weeks, and avacincaptad pegol (Izervay; Astellas Pharma), a C5 inhibitor given every 4 weeks, both achieved moderate reductions in lesion growth rates of approximately 14% to 28% over 2 years.23-25 However, neither therapy halts disease progression or improves visual function, and both require frequent intravitreal injections that impose significant treatment burden on elderly patients.
More concerning, both approved therapies increased the incidence of conversion to neovascular AMD. The European Medicines Agency (EMA) refused marketing authorization for pegcetacoplan based on an unfavorable overall benefit-risk profile (did not demonstrate meaningful improvement in patients’ vision or everyday functioning). Likewise, the EMA rendered a similar provisional opinion on avacincaptad pegol, and the application was withdrawn. These limitations highlight the need for more comprehensive therapeutic approaches that address multiple pathologic mechanisms while reducing treatment burden and minimizing adverse effects.
These current approved therapies follow single-pathway approaches, targeting either C3 or C5 complement components. Although this provides some benefit through complement modulation, it fails to address other critical aspects of GA pathogenesis, including direct neuroprotection, oxidative stress, and cellular survival mechanisms. The modest efficacy observed with these approaches suggests that GA’s complex pathophysiology requires multitarget therapeutic strategies.
The Bicistronic Gene Therapy Approach
IKAR-001 represents a novel dual pathway gene therapy with a bicistronic design that addresses GA’s multifaceted pathophysiology through simultaneous expression of 2 therapeutic proteins: PEDF and soluble CD46 (sCD46). This dual-mechanism strategy targets both neuroprotection and complement regulation, potentially providing superior efficacy compared to single-pathway approaches.
The therapy uses an adeno-associated virus serotype 8 (AAV8) vector, chosen for its excellent tropism for RPE cells and established safety profile in ocular applications. The bicistronic design enables the expression of 2 synergistic therapeutic proteins to be expressed from a single vector. A constitutive promoter drives robust expression and an optimised furin/viral 2A linker ensures correct protein processing and independent secretion of each protein.
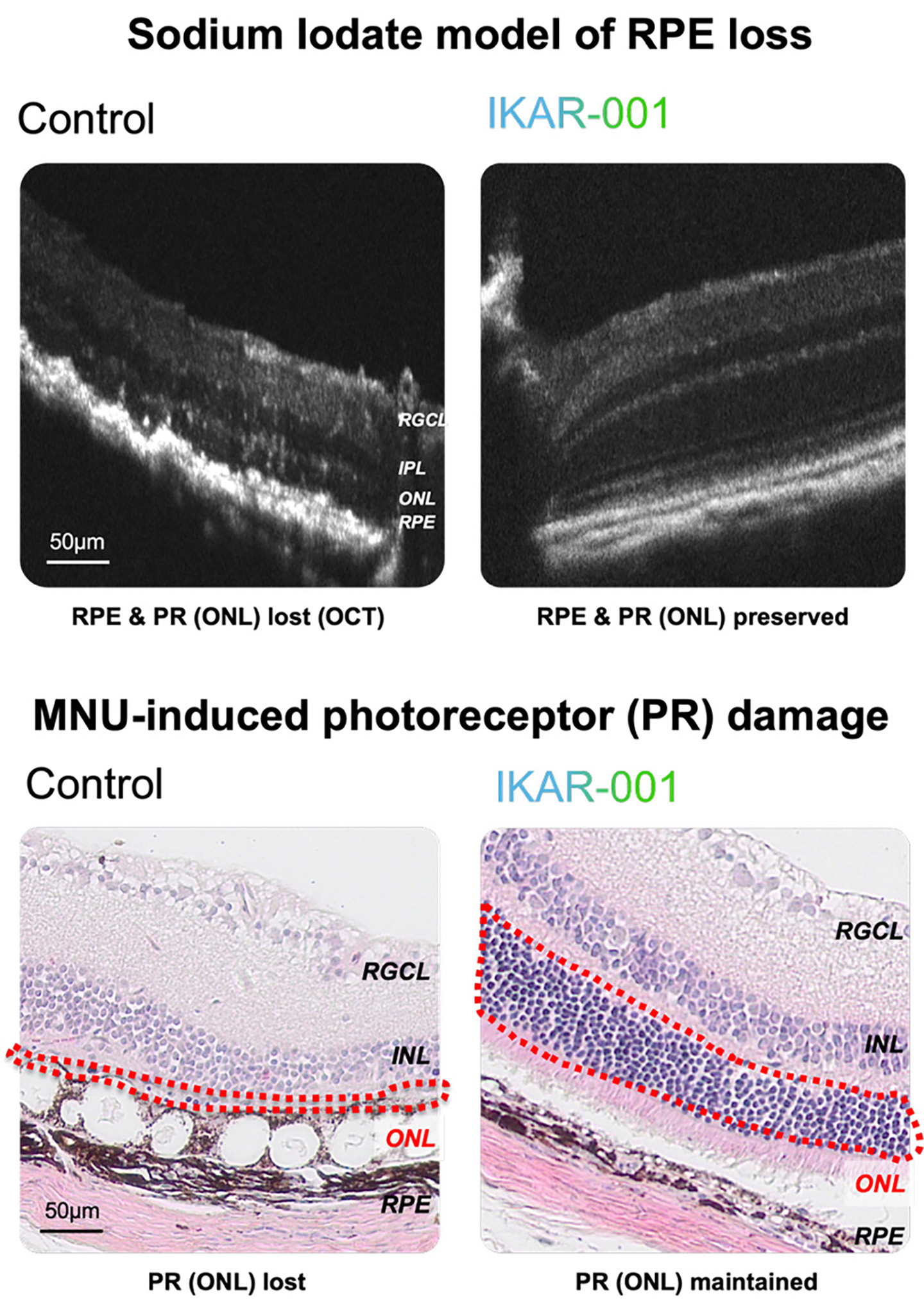
Figure 3. IKAR-001 preserves retinal structure in preclinical models of retinal degeneration. In a sodium iodate model of retinal pigment epithelium (RPE) toxicity, optical coherence tomography demonstrates loss of RPE and photoreceptor layers (ONL) in control eyes, while IKAR-001-treated eyes show preserved retinal architecture. In a MNU-induced photoreceptor damage model, histologic sections reveal near-complete loss of the outer nuclear layer (ONL, red dashed outline) in control eyes, whereas IKAR-001 treatment maintains photoreceptor cell density. Retinal layers labeled: RGCL, retinal ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium.
Subretinal administration offers several important advantages. Direct delivery to target RPE and photoreceptor cells maximizes therapeutic efficiency while minimizing systemic exposure. The anatomic isolation of the subretinal space reduces systemic exposure and the risk of systemic immune responses.26 Most importantly, gene therapy provides sustained local expression with a single injection, potentially offering years of therapeutic benefit compared to the frequent injections required by current treatments.
Importantly, as detailed above, both PEDF and sCD46 are naturally occurring proteins in the eye that become downregulated as GA disease progresses. Compared to gene therapies that produce non-naturally occurring proteins like ranibizumab or aflibercept, there may be a lower risk of immunogenic reactions. This replacement therapy rationale, restoring proteins that are depleted early in disease, represents a physiologically grounded approach to treating GA.
Preclinical Supporting Evidence Dual-Pathway Therapy
Extensive preclinical studies have demonstrated IKAR-001’s superior efficacy compared to single-component approaches. In complement modulation models using primary human cells and ex vivo systems, the bicistronic vector showed superior inhibition of membrane attack complex (MAC) formation compared to individual PEDF or sCD46 components. Unlike approved complement inhibitors that use 1:1 stoichiometric binding, sCD46 acts catalytically with complement factor I to inactivate complement components, a potentially more efficient mechanism. Notably, the bicistronic combination demonstrated synergistic rather than merely additive effects; although PEDF alone had no impact on complement modulation in isolated cell systems, the combination substantially outperformed either component alone in models of complex retinal inflammation.
Retinoprotective studies using sodium iodate, MNU, and NMDA-induced damage models demonstrated preserved retinal function and reduced cell death. Importantly, these studies showed protection across multiple retinal cell types, including RPE cells, photoreceptors, and ganglion cells, suggesting broad neuroprotective effects. The preservation of retinal function, as measured by electroretinography, indicates that the therapy may not only slow anatomic progression but also maintain visual function, a critical endpoint that current therapies have failed to achieve.
Head-to-head comparisons with complement inhibitors, including vectors expressing CR1, sCD59, or CFI, demonstrated superior efficacy for the IKAR-001 bicistronic approach in laser CNV models across multiple endpoints including MAC area, lesion area, and vascular permeability. This suggests that the specific combination of PEDF and sCD46 provides unique therapeutic advantages beyond what might be achieved with other complement regulatory proteins alone.
Nonhuman primate studies have demonstrated that IKAR-001 is safe and well tolerated following subretinal administration, with robust transduction of RPE and photoreceptors and functionally active therapeutic protein levels achieved in the vitreous. These findings, combined with the established clinical precedent for subretinal AAV gene therapy demonstrated by Luxturna (voretigene neparvovec-rzyl; Spark Therapeutics) and RGX-314 (Regenxbio), support the translational potential of this approach.
Precedent for Ocular Gene Therapy
Recent clinical developments have established strong precedents for the IKAR-001 approach. Luxturna, the FDA-approved subretinal AAV2 gene therapy for RPE65-associated inherited retinal dystrophy, demonstrated the safety and regulatory pathway for subretinal AAV gene delivery.27 Recent phase 1/2 data for RGX-314, a subretinal AAV8 gene therapy for neovascular AMD, showed generally well-tolerated delivery with sustained protein expression and no clinically recognized immune responses.28
Previous clinical experience with dual-pathway approaches has also demonstrated feasibility. The GEM study showed safe subretinal delivery of dual antiangiogenic proteins using a lentiviral vector to co-express endostatin and angiostatin, with sustained expression over multiple years and well-tolerated procedures.29 These precedents collectively derisk the regulatory and clinical pathways for dual-pathway gene therapy approaches.
Platform Extension Opportunities
The dual-pathway gene therapy platform offers significant expansion opportunities beyond GA. The dual-pathway mechanism targeting neuroinflammation and retinal cell death addresses common pathologic mechanisms across retinal degenerations. The gene-agnostic neuroprotective approach makes IKAR-001 applicable to retinitis pigmentosa (RP), potentially addressing the vast majority of patients regardless of specific genetic mutations. This represents a substantial improvement over current gene-specific therapies that treat fewer than 5% of RP patients. The approximately 1.5 million RP patients worldwide represent a significant unmet need that could benefit from this approach.
Ikarovec’s pipeline includes additional programs: IKAR-002 for wet AMD, combining anti-VEGF and antifibrotic mechanisms to address the fibrosis that develops in approximately half of wet AMD patients at 24 months despite anti-VEGF therapy, and IKAR-003 for intermediate AMD, potentially providing earlier intervention before irreversible damage occurs.
Looking Forward
The success of dual-pathway approaches in GA treatment could establish new therapeutic paradigms for retinal degenerative diseases. Unlike current therapies that modestly slow progression without functional benefit, the combination of neuroprotection and complement modulation may achieve anatomic and functional disease stabilization or even functional improvement, particularly at the junctional zones of geographic atrophy. The potential for sustained therapeutic benefit with single administration addresses a critical unmet need in GA treatment, reducing treatment burden for elderly patients while potentially improving efficacy.
The regulatory landscape for neuroprotective retinal therapies has evolved significantly. In March 2025, the FDA approved Encelto (Neurotech Pharmaceuticals), an encapsulated cell therapy delivering ciliary neurotrophic factor, for macular telangiectasia type 2. This approval represents several important precedents: the first neuroprotective therapy approved for a macular degenerative disease, with a primary endpoint of photoreceptor protection as measured by OCT, and, as a form of gene-modified cell therapy, further validation of genetic medicine approaches for retinal disease.
The integration of neuroprotection and complement modulation represents a rational therapeutic approach based on our evolving understanding of GA pathophysiology. By simultaneously targeting complement dysregulation and providing direct neuroprotection, dual-pathway gene therapy approaches offer the promise of more effective, convenient, and comprehensive treatment for one of the most challenging causes of age-related blindness. RP

References
1. American Macular Degeneration Foundation. Geographic atrophy—advanced form of dry macular degeneration. Accessed February 2, 2026. https://www.macular.org/about-macular-degeneration/geographic-atrophy
2. Gao J, Liu RT, Cao S, et al. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediators Inflamm. 2015;2015:690243. doi:10.1155/2015/690243
3. Marneros AG. Role of inflammasome activation in neovascular age-related macular degeneration. FEBS J. 2023;290(1):28-36. doi:10.1111/febs.16278
4. West EE, Woodruff T, Fremeaux-Bacchi V, Kemper C. Complement in human disease: approved and up-and-coming therapeutics. Lancet. 2024;403(10424):392-405. doi:10.1016/S0140-6736(23)01524-6
4. Harris CL, Pouw RB, Kavanagh D, Sun R, Ricklin D. Developments in anti-complement therapy; from disease to clinical trial. Mol Immunol. 2018;102:89-119. doi:10.1016/j.molimm.2018.06.0084
5. Khan AH, Chowers I, Lotery AJ. Beyond the complement cascade: insights into systemic immunosenescence and inflammaging in age-related macular degeneration and current barriers to treatment. Cells. 2023;12(13):1708. doi:10.3390/cells12131708
6. Vogt SD, Curcio CA, Wang L, et al. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp Eye Res. 2011;93(4):413-423. doi:10.1016/j.exer.2011.06.002
7. Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29(2):95-112. doi:10.1016/j.preteyeres.2009.11.003
8. Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J Pathol. 2013;229(5):729-742. doi:10.1002/path.4128
9. Lyzogubov VV, Bora PS, Wu X, et al. The complement regulatory protein CD46 deficient mouse spontaneously develops dry-type age-related macular degeneration-like phenotype. Am J Pathol. 2016;186(8):2088-2104. doi:10.1016/j.ajpath.2016.03.021
10. Lyzogubov V, Wu X, Jha P, et al. Complement regulatory protein CD46 protects against choroidal neovascularization in mice. Am J Pathol. 2014;184(9):2537-2548. doi:10.1016/j.ajpath.2014.06.001
11. Sweigard JH, Cashman SM, Kumar-Singh R. Adenovirus-mediated delivery of CD46 attenuates the alternative complement pathway on RPE: implications for age-related macular degeneration. Gene Ther. 2011;18(6):613-621. doi:10.1038/gt.2011.6
12. Machalińska A, Safranow K, Mozolewska-Piotrowska K, Dziedziejko V, Karczewicz D. PEDF and VEGF plasma level alterations in patients with dry form of age-related degeneration—a possible link to the development of the disease. Klin Oczna. 2012;114(2):115-120.
13. Bhutto IA, McLeod DS, Hasegawa T, et al. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82(1):99-110. doi:10.1016/j.exer.2005.05.007
14. Warner EF, Vaux L, Boyd K, Widdowson PS, Binley KM, Osborne A. Ocular delivery of Pigment Epithelium-Derived Factor (PEDF) as a neuroprotectant for geographic atrophy. Aging Dis. 2024;15(5):2003-2007. doi:10.14336/AD.2024.0216-1
15. Wang X, Liu X, Ren Y, et al. PEDF protects human retinal pigment epithelial cells against oxidative stress via upregulation of UCP2 expression. Mol Med Rep. 2019;19(1):59-74. doi:10.3892/mmr.2018.9645
16. Wang Y, Subramanian P, Shen D, Tuo J, Becerra SP, Chan CC. Pigment epithelium-derived factor reduces apoptosis and pro-inflammatory cytokine gene expression in a murine model of focal retinal degeneration. ASN Neuro. 2013;5(5):e00126. doi:10.1042/AN20130028
17. Rebustini IT, Crawford SE, Becerra SP. PEDF Deletion induces senescence and defects in phagocytosis in the RPE. Int J Mol Sci. 2022;23(14):7745. doi:10.3390/ijms23147745
18. Bernardo-Colón A, Dong L, Abu-Asab M, Brush RS, Agbaga MP, Becerra SP. Ablation of pigment epithelium-derived factor receptor (PEDF-R/Pnpla2) causes photoreceptor degeneration. J Lipid Res. 2023;64(5):100358. doi:10.1016/j.jlr.2023.100358
19. Bernardo-Colón A, Bighinati A, Parween S, et al. H105A peptide eye drops promote photoreceptor survival in murine and human models of retinal degeneration. Commun Med (Lond). 2025;5(1):81. doi:10.1038/s43856-025-00789-8
20. Holekamp NM, Bouck N, Volpert O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am J Ophthalmol. 2002;134(2):220-227. doi:10.1016/s0002-9394(02)01549-0
21. Shughoury A, Boucher N, Aggarwal N, Ciulla TA. Three-year clinical outcomes in geographic atrophy: an analysis of 18,712 patient eyes. Retina. 2025;45(2):188-197. doi:10.1097/IAE.0000000000004285
22. Liao DS, Grossi FV, El Mehdi D, et al. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology. 2020;127(2):186-195. doi:10.1016/j.ophtha.2019.07.011
23. Syfovre (pegcetacoplan injection) prescribing information. Apellis Pharmaceuticals, Inc; 2023. Accessed February 2, 2026. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217171s002lbl.pdf
24. Izervay (avacincaptad pegol intravitreal solution) prescribing information. Astellas Pharma US, Inc; 2023. Accessed February 2, 2026. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/217225s001lbl.pdf
25. Seitz IP, Michalakis S, Wilhelm B, et al. Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of AAV8 in nonhuman primates. Invest Ophthalmol Vis Sci. 2017;58(13):5792-5801. doi:10.1167/iovs.17-22473
26. Luxturna (voretigene neparvovec-rzyl) prescribing information. Spark Therapeutics, Inc; May 2022. Accessed February 2, 2026. https://www.fda.gov/media/109906/download
27. Campochiaro PA, Avery R, Brown DM, et al. Gene therapy for neovascular age-related macular degeneration by subretinal delivery of RGX-314: a phase 1/2a dose-escalation study. Lancet. 2024;403(10436):1563-1573. doi:10.1016/S0140-6736(24)00310-6
28. Campochiaro PA, Lauer AK, Sohn EH, et al. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum Gene Ther. 2017;28(1):99-111. doi:10.1089/hum.2016.117