







Inherited retinal diseases (IRDs) are a group of conditions responsible for irreversible vision loss in 20% to 25% of working-age individuals globally.1-3 Although retina specialists encounter IRDs with increasing frequency, at this time there is only one FDA-approved therapy across all IRDs. There is hope, however, in a specific type of gene therapy called optogenetics. This mutation- and disease-agnostic approach introduces genes encoding for opsins, or light-sensitive proteins, into the retina to regain light sensitivity and restore a degree of functional vision.4
In my practice, the most common IRDs we see are retinitis pigmentosa (RP), characterized by retinal pigmentary changes and a gradual loss of both night and peripheral vision, and Stargardt disease, characterized by yellow flecks in the macula and retinal midperiphery progressing to macular beaten bronze atrophic changes. Patients with RP can progress to complete blindness,5 and those with Stargardt disease typically experience debilitating central vision loss6 and severe difficulty with daily tasks, including reading and identifying faces.7
One of the most heartbreaking aspects of treating patients with IRDs is that they often present with advanced disease. RP typically begins with subtle or undetectable symptoms like night blindness and peripheral vision loss that can go unrecognized for years. It’s not uncommon for patients to receive a diagnosis in their 30s or 40s, only to realize their initial symptoms started in adolescence. Patients with Stargardt disease have more variable presentation, with some presenting earlier and others presenting later, but both conditions follow a similar relentless course.
An Evolution of Treatment Approaches
It can be emotional for patients to hear, “There’s nothing we can do to save your vision.” We might advise them to wear sunglasses, avoid excessive sunlight, and eat a healthy diet, but these lifestyle suggestions are not medical therapy. Vitamin A supplementation, once believed to slow RP progression,8 is no longer supported by recent data.9 In the case of Stargardt disease, high levels of vitamin A can even be harmful.10
Diagnosis and treatments for IRDs are gradually evolving. Genetic testing is more accessible now, allowing us to identify specific mutations and understand the molecular basis of the disease. Nevertheless, most current treatments are highly targeted and gene specific. Voretigene neparvovec-rzyl (Luxturna; Spark Therapeutics), for example, was the first approved gene therapy, but it is only indicated for Leber congenital amaurosis (LCA) secondary to biallelic RPE65 mutations, which is one of hundreds of genetically characterized IRDs. Although the approval of Luxturna, which is designed to replace a defective gene with a functioning one using an adeno-associated viral (AAV) vector, introduced a new era in gene therapy, it only helps a small percentage of the broader IRD population. In the United States, between 1,000 and 2,000 individuals have LCA.11
The Era of Optogenetics
Traditional gene replacement therapy is designed to preserve rather than restore vision. It works best when an eye has viable photoreceptors, encoding for functional proteins to prevent photoreceptor loss. If a patient presents after most of their photoreceptors have degenerated, traditional gene therapy may have reduced efficacy.
Optogenetics offers a potential groundbreaking shift, especially for patients with late-stage disease. It uses remaining intact bipolar and ganglion cells to express light-sensitive opsin proteins, allowing these downstream neurons to respond to visual stimuli and essentially act as new light sensors, even in the absence of functioning rods and cones. The hope is that optogenetics can help patients regain light sensitivity and restore a degree of visual function to areas of the retina that have already lost photoreceptors and RPE cells.4
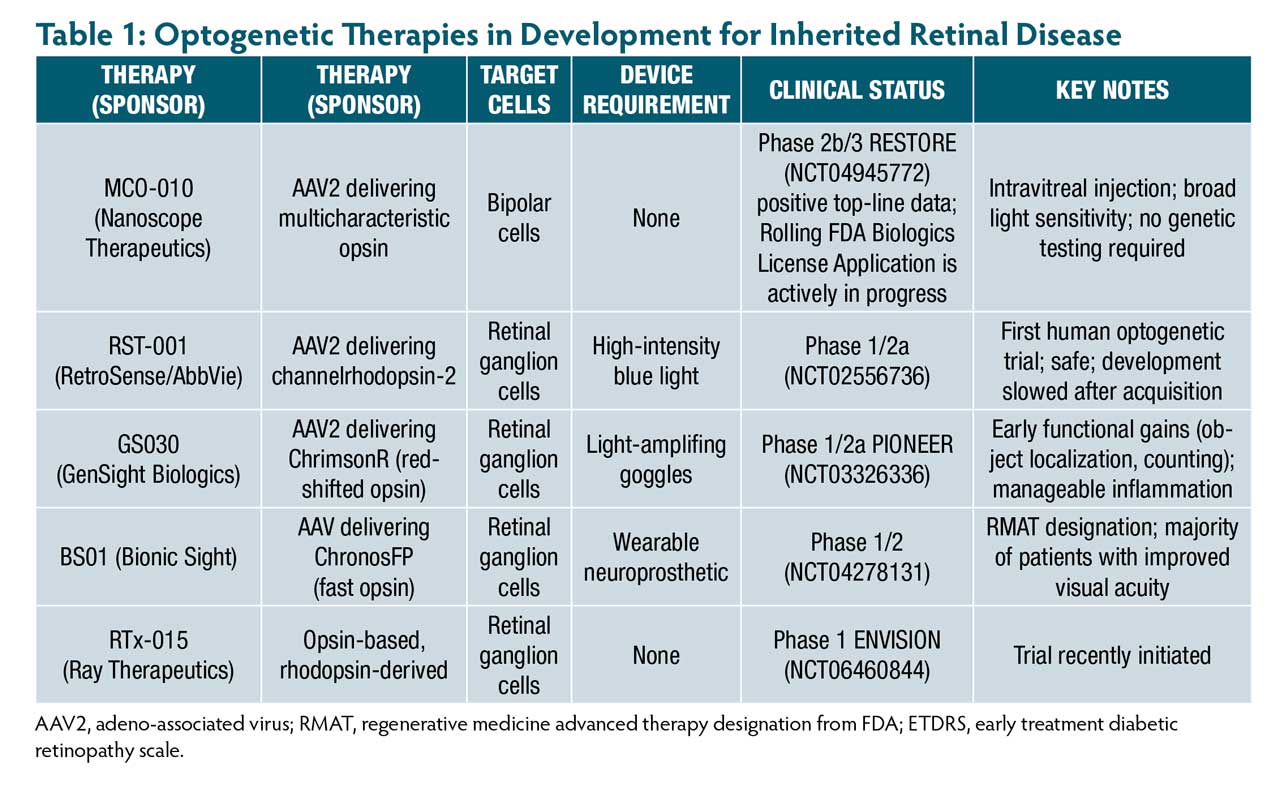
An approved optogenetics therapy would significantly expand the therapeutic window for IRDs. Several optogenetics therapies are in development (Table 1), including MCO-010 (Nanoscope Therapeutics), RST-001 (RetroSense/Allergan), GS030 (GenSight), BS01 (Bionic Sight), and RTx-015 (Ray Therapeutics).
One of these optogenetic therapies in development, MCO-010, delivers the multicharacteristic opsin transgene by a proprietary adeno-associated virus (AAV2) as a one-time, in-office intravitreal injection for gene- agnostic RP.12-15 MCO-010 targets bipolar cells to express the MCO-010 transmembrane ion channel protein. When ambient light hits the retina, the MCO-010 protein undergoes a conformational change, opening the ion channel and depolarizing the cell, thereby triggering electrical signals that are transferred through the visual pathway to the brain. MCO-010 protein is sensitive across the visible light spectrum. It does not require a light-intensifying device for opsin activation or to mimic photoreceptor function.
RESTORE, a phase 2b/3 randomized, controlled, multicenter clinical trial, showed positive 2-year results with MCO-010 to restore vision in patients with severe vision loss from RP.16 Patients received 1 of 2 doses of the drug or a sham treatment. At week 52, 40% of patients receiving either high-dose or low-dose MCO-010 experienced a mean improvement in BCVA from baseline of at least 0.3 logMAR (3 lines or 15-letters ETDRS equivalent), which were maintained out to 152 weeks.
Due to their mechanism of action, optogenetic therapies are theoretically applicable to other diseases across the IRD spectrum. For example, MCO-010 induces de novo photosensitivity of intact inner retina neurons in settings of advanced photoreceptor loss and is theoretically applicable to other IRDs like Stargardt disease. Early clinical data from 6 patients with severe vision loss from Stargardt disease showed a positive safety profile and functional improvements.17
Other promising optogenetic treatments in development, such as RTx-015, BS01, and GS030, target retinal ganglion cells for the treatment of RP, Stargardt disease, and geographic atrophy. Ray Therapeutics is also conducting late-stage preclinical studies of RTx-021, which targets bipolar cells.
Moving Beyond Lifestyle Advice
If optogenetics proves successful in rigorous clinical trials and demonstrates both efficacy and safety, we may be able to offer patients with severe disease more than monitoring progression, offering low-vision support, and providing lifestyle advice. My patients with end-stage disease long to regain independence through functional vision. I think of those who return year after year, hopeful yet hearing the same words: “Everything looks stable.” For them, optogenetics could represent a new outlook on life.
Should optogenetic therapy ultimately gain regulatory approval, several important considerations will follow. First, patient selection criteria must be clearly defined and aligned with labeling to ensure that candidates most likely to benefit are identified. Second, physicians must guard against the temptation to overpromise and underdeliver, particularly with vulnerable patients who are often desperate for hope. Third, clinical workflows may need to adapt to address patient expectations and coordinate post-treatment monitoring.
Conclusion
While traditional gene therapy focuses on preserving patients’ remaining vision, optogenetics aims to restore vision that has already been lost. Retina specialists should remain cautiously optimistic, with the hope that this approach may soon help redefine the treatment paradigm for our IRD patients. RP

References
1. Science Direct. Stem cell therapy and retinal regeneration. Accessed September 7, 2025. https://www.sciencedirect.com/topics/neuroscience/retinal-degeneration
2. Crewe JM, Morlet N, Morgan WH, et al. Mortality and hospital morbidity of working-age blind. Br J Ophthalmol. 2013;97(12):1579-1585. doi:10.1136/bjophthalmol-2013-303993
3. Liew G, Michaelides M, Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open. 2014;4(2):e004015. doi:10.1136/bmjopen-2013-004015
4. Wood EH, Kreymerman A, Kowal T, et al. Cellular and subcellular optogenetic approaches towards neuroprotection and vision restoration. Prog Retin Eye Res. 2023;96:101153. doi:10.1016/j.preteyeres.2022.101153
5. Mayo Clinic. Diagnosis, clinical trials and treatments for inherited retinal disease. October 14, 2023. Accessed June 23, 2025. https://www.mayoclinic.org/medical-professionals/ophthalmology/news/diagnosis-clinical-trials-and-treatments-for-inherited-retinal-diseases/mac-20556115
6. National Eye Institute. Stargardt disease. December 4, 2024. Accessed September 7, 2025. https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/stargardt-disease
7. Royal National Institute of Blind People. Stargardt disease. February 1, 2024. Accessed September 7, 2025. https://www.rnib.org.uk/your-eyes/eye-conditions-az/stargardt-disease
8. Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022
9. Comander J, Weigel DiFranco C, Sanderson K, et al. Natural history of retinitis pigmentosa based on genotype, vitamin A/E supplementation, and an electroretinogram biomarker. JCI Insight. 2023;8(15):e167546. doi:10.1172/jci.insight.167546
10. Radu RA, Yua Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49(9):3821-3829. doi:10.1167/iovs.07-1470
11. Hu ML, Edwards TL, O’Hare F, et al. Gene therapy for inherited retinal diseases: progress and possibilities. Clin Exp Optom. 2021;104(4):444-454. doi:10.1080/08164622.2021.1880863
12. Wright WW, Gajjeraman S, Batabyal S, et al. Restoring vision in mice with retinal degeneration using multicharacteristic opsin. Neurophotonics. 2017;4(4):041505. doi:10.1117/1.NPh.4.4.041505
13. Batabyal S, Gajjeraman S, Pradhan S, Bhattacharya S, Wright W, Mohanty S. Sensitization of ON-bipolar cells with ambient light activatable multi-characteristic opsin rescues vision in mice. Gene Ther. 2021;28(3-4):162-176. doi:10.1038/s41434-020-00200-2
14. Batabyal S, Kim S, Carlson M, et al. Multi-characteristic opsin therapy to functionalize retina, attenuate retinal degeneration, and restore vision in mouse models of retinitis pigmentosa. Transl Vis Sci Technol. 2024;13(10):25. doi:10.1167/tvst.13.10.25
15. Mohanty SK, Mahapatra S, Batabyal S, et al. A synthetic opsin restores vision in patients with severe retinal degeneration. Molecular Therapy. 2025;33(5):2279–2290. doi:10.1016/j.ymthe.2025.03.031
16. Nanoscope presented positive 2-year randomized, controlled trial results of MCO-010 for retinitis pigmentosa. News release. October 31, 2024. Accessed June 30, 2025. https://nanostherapeutics.com/2024/10/31/nanoscope-presented-positive-2-year-randomized-controlled-trial-results-of-mco-010-for-retinitis-pigmentosa/
17. Nanoscope Therapeutics announces completion of enrollment in STARLIGHT phase 2 clinical trial of MCO-010 optogenetic gene therapy for Stargardt disease. News release. September 13, 2022. Accessed September 10, 2025. https://nanostherapeutics.com/2022/09/13/nanoscope-therapeutics-announces-completion-of-enrollment-in-starlight-phase-2-clinical-trial-of-mco-010-optogenetic-gene-therapy-for-stargardt-disease/