







Stargardt disease is among the most prevalent forms of inherited macular degeneration, affecting approximately 1 in 10,000 individuals.1,2 This progressive disorder often leads to loss of central vision, significantly impacting daily activities. It is named after the German ophthalmologist Karl Stargardt, who first described the condition in 1901. Understanding the clinical presentation and underlying pathology of Stargardt disease is crucial for identifying patients as well as developing and offering effective treatments. Ongoing research is making strides toward potential therapeutic interventions that could alter the course of the disease.
Molecular Mechanisms and Pathophysiology of Stargardt Disease
Stargardt disease is caused by variants in the ABCA4 gene, a member of the superfamily of ATP-binding cassette (ABC) transporters, which plays a critical role in the visual cycle.3 The visual cycle begins when photons of light enter the eye and are absorbed by photoreceptor cell opsins. This absorption causes a change in the structure of the photopigments (eg, opsin proteins), leading to the isomerization of 11-cis-retinal to all-trans-retinal.4 In a normal functioning retina, the ABCA4 protein aids in the efficient removal of all-trans-retinal from photoreceptors, preventing the accumulation of toxic byproducts.3,5 However, ABCA4 variants disrupt this process, leading to the buildup of all-trans-retinal and its condensation product, N-retinylidene-N-retinylethanolamine (A2E).
A2E, a vitamin A dimer, is a component of lipofuscin, a fluorescent pigment that accumulates within the retinal pigment epithelium (RPE) cells over time in disease states such as Stargardt disease.6 The accumulation of A2E and other toxic byproducts in the RPE cells leads to oxidative stress, inflammation, and eventually cell death. As the RPE cells degenerate, the overlying photoreceptors also begin to die, particularly in the macula.
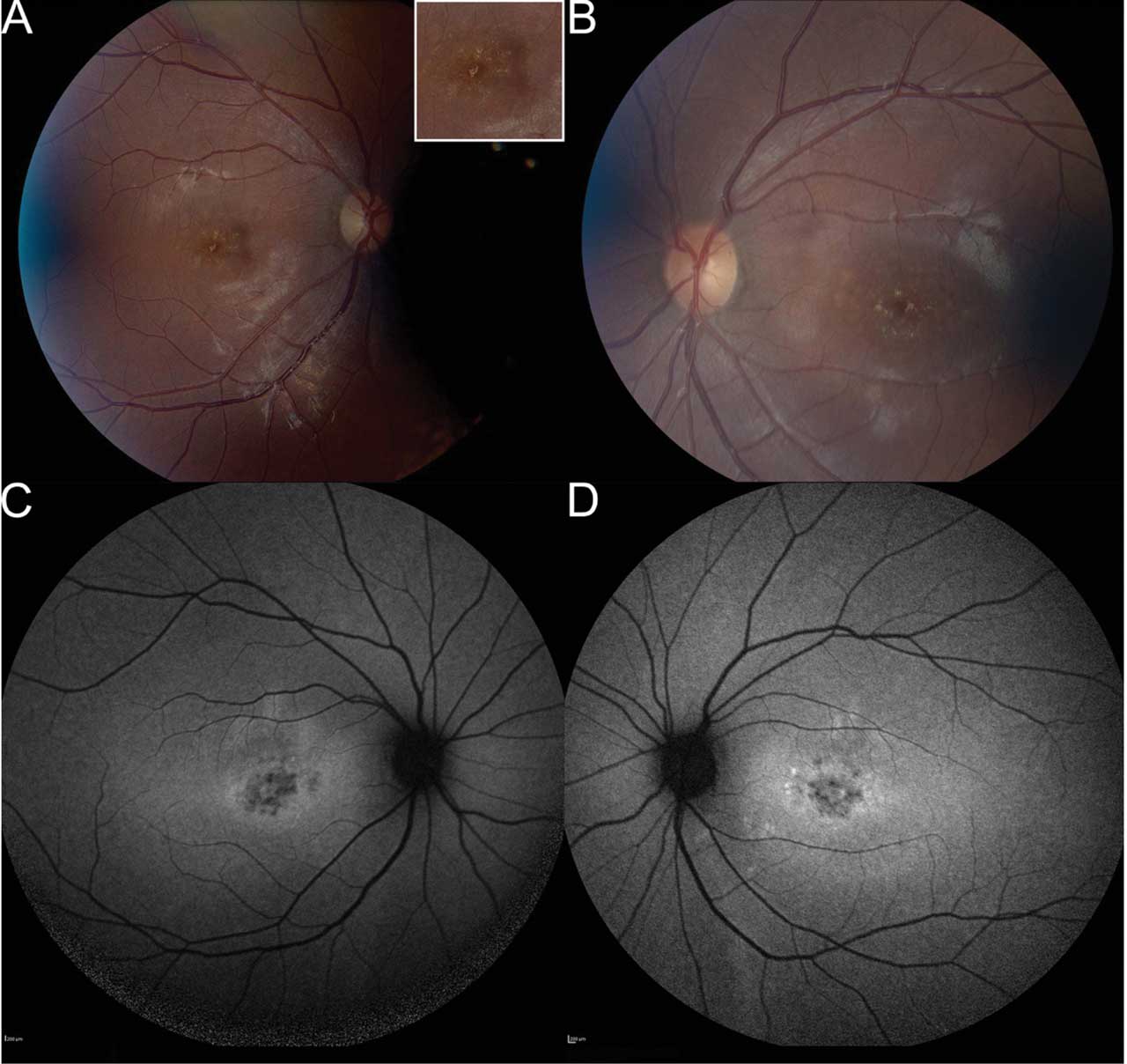
Figure 1. Fundus photography of a 20-year-old patient, who presented with a history of central vision loss starting in her mid to late teens. Right fundus (A) shows pisciform lipofuscin deposits concentrated parafoveally with normal appearance of nerve, vessels, and mid-periphery. Inset demonstrates higher magnification of the lipofuscin deposits. Left eye (B) shows symmetry with right eye, including parafoveal pisciform lipofuscin deposits. Fundus autofluorescence (FAF) of the right (C) and left (D) eyes demonstrates symmetric flecks that are mostly hypoautofluorescent. There are a few hyperautofluorescent lesions parafoveally in left eye. There is a ring of hyperautofluorescence in both eyes around parafoveal lesions. There are no lesions outside the central macula. Microperimetry showed decentered fixation and preferred retinal locus in the right eye with decreased central sensitivity in both eyes (refer to Figure 4). Stargardt disease genetic testing identified 3 pathogenic variants in the ABCA4 gene, specifically homozygous for ABCA4 c.5882G>A (p.Gly1967Glu), which was pathogenic, and heterozygous for ABCA4 c.634C>T (p.Arg212Cys), which was pathogenic. All images courtesy Brittni A. Scruggs, MD, PhD.
Clinical Presentation and Diagnosis
The clinical presentation of Stargardt disease can vary widely, depending on the specific variants in the ABCA4 gene and the age of onset.7 In classic Stargardt disease, the disease manifests in childhood or adolescence (Figures 1 and 4), with patients reporting difficulty in reading, recognizing faces, or performing tasks that require fine visual detail. However, some individuals may not experience symptoms until later in life (Figures 2 and 3), and the rate of disease progression can vary. In a study by Fishman et al, the authors noted the likelihood of retaining a visual acuity of 20/40 or better in at least 1 eye was 52% by age 19, 32% by age 29, and 22% by age 39.8 Also, when a patient’s visual acuity fell below 20/40, it typically declined quickly and then stabilized at 20/200.
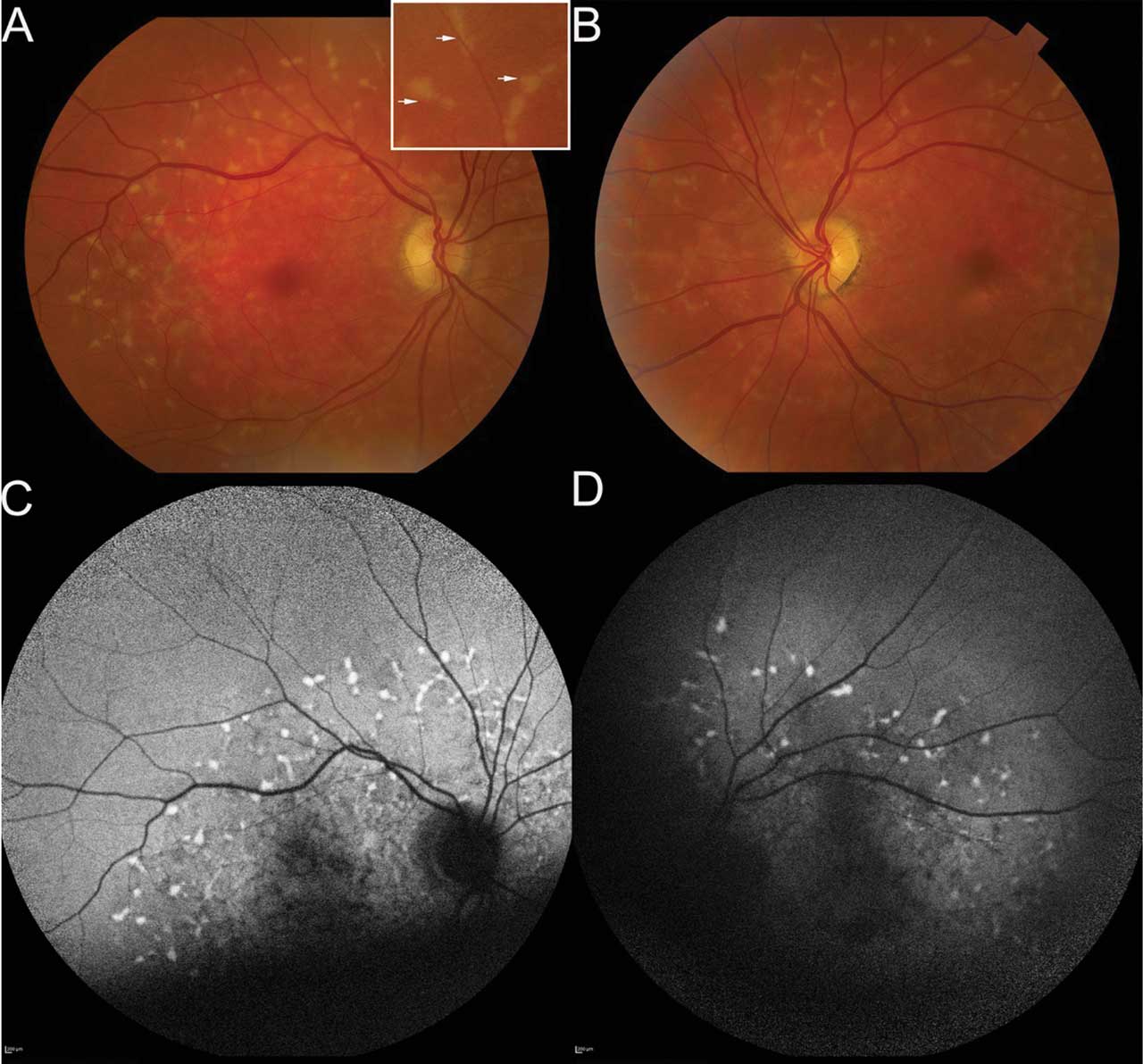
Figure 2. Fundus photography of a 72-year-old patient who presented with onset of blurred vision and night blindness; his corrected visual acuity was 20/60 in both eyes. The right fundus photo (A) shows pisciform flecks throughout the perifoveal region and extending into mid-periphery. Inset shows higher magnification of these lipofuscin deposits. Left fundus photo (B) shows findings similar to the right eye, with an additional finding of paracentral geographic atrophy inferior and temporal to the fovea. Fundus autofluorescence (FAF) imaging of right (C) and left (D) eyes demonstrates hyperautofluorescent flecks, each with subtle hypoautofluorescent ring. This is hypoautofluorescence inferior to fovea in left eye in area of atrophy. Quality of FAF was limited by patient’s poor fixation. Microperimetry (not shown) demonstrated fixation in the superior macula in both eyes with decreased sensitivity centrally. Genetic testing demonstrated the patient was heterozygous for ABCA4 c.5461-10T>C, which was pathogenic, and homozygous for ABCA4 c.5603A>T, p.(Asn1868Ile), which was a risk factor. This confirmed the diagnosis of late-onset Stargardt disease.
The diagnosis of Stargardt disease is based on a combination of clinical findings, imaging studies, and genetic testing. Key diagnostic techniques include the fundus examination, fundus autofluorescence (FAF), optical coherence tomography (OCT), and genetic testing. A detailed fundus examination of the retina may exhibit a characteristic “beaten bronze” appearance in the macula, along with the presence of yellowish-white flecks or deposits in the retina. These deposits, often called pisciform flecks, can resemble that of fish tails (hence, pisces) with a 120° angle between 2 adjacent deposits. Patients may have a normal-appearing fundus, however, necessitating other means of testing. OCT may reveal overall outer retinal thinning of the central and/or paracentral macula due to loss of the photoreceptor layer and disruption of the RPE.9 In addition, external limiting membrane thickening has been noted in patients with Stargardt disease.10 FAF imaging is particularly useful for visualizing lipofuscin accumulation in the RPE.11 Areas of increased autofluorescence (hyperautofluorescence) generally correspond to lipofuscin deposits, whereas areas of decreased autofluorescence (hypoautofluorescence) often indicate RPE atrophy, often correlating to scotoma.
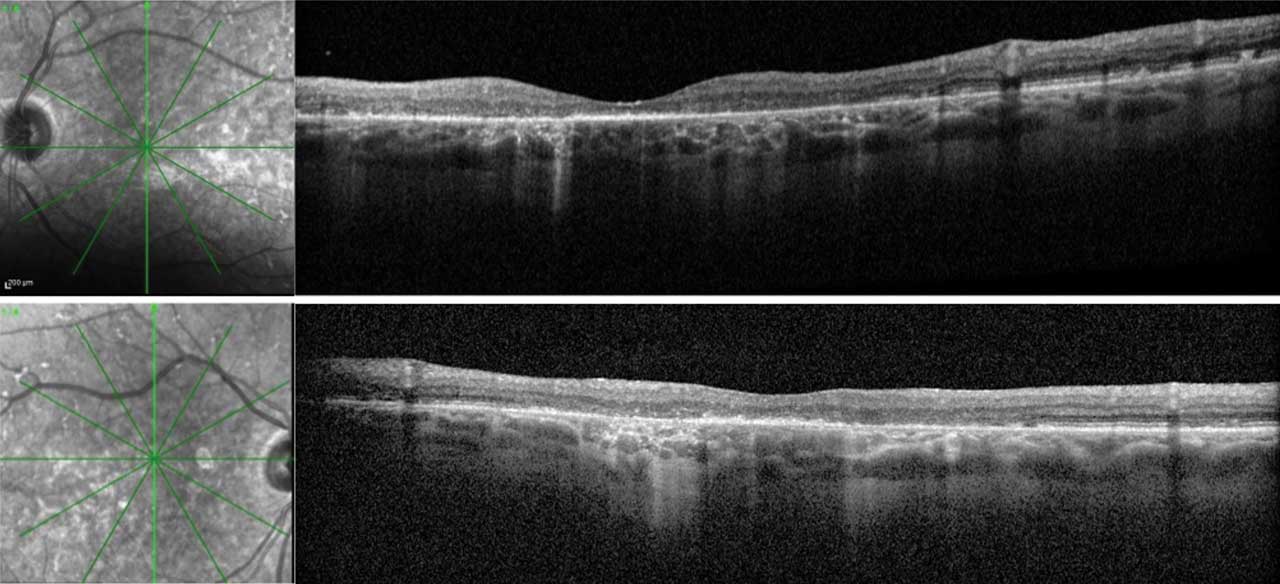
Figure 3. Optical coherence tomography (OCT) of both eyes in a patient with late-onset Stargardt disease. Right eye (upper) and left eye (lower) images include vertical cross-sectional OCT images with their respective infrared images. This patient is also represented in Figure 2. The RPE deposits throughout the macula with a predominance in the perifoveal region are consistent with the pisciform lesions clinically seen in both eyes. There is patchy outer retinal loss with resultant choroidal hypertransmission defects in both eyes. A small area of preserved ellipsoid zone centrally in both eyes allows the patient to have functional (20/60) vision in both eyes.
Electroretinography (ERG) can aid in the diagnosis or prognosis of Stargardt disease. Using ERG, Lois et al showed that 3 different subgroups exist, which correlate to disease severity.12 The first group demonstrates severe pattern ERG abnormality with normal scotopic and full-field ERGs. The second group demonstrated loss of photopic function. Finally, the third group demonstrated both cone and rod dysfunction on full-field ERG. Because ERG testing requires bright flashes that can contribute to lipofuscin accumulation, serial ERG testing should be avoided for patients with suspected or confirmed Stargardt disease when possible.13,14 Functional tests include microperimetry (Figure 4) and Humphrey visual field; these help with prognosis and monitoring for progression. Microperimetry is an important tool in clinical trial design, as highlighted in the results of the RUSH2A study15 and recent panel discussion at ARVO 2024.16 This tool can capture changes in a small area in the retina that might be the only area demonstrating change over the relatively short 2-year to 3-year period of a clinical trial.
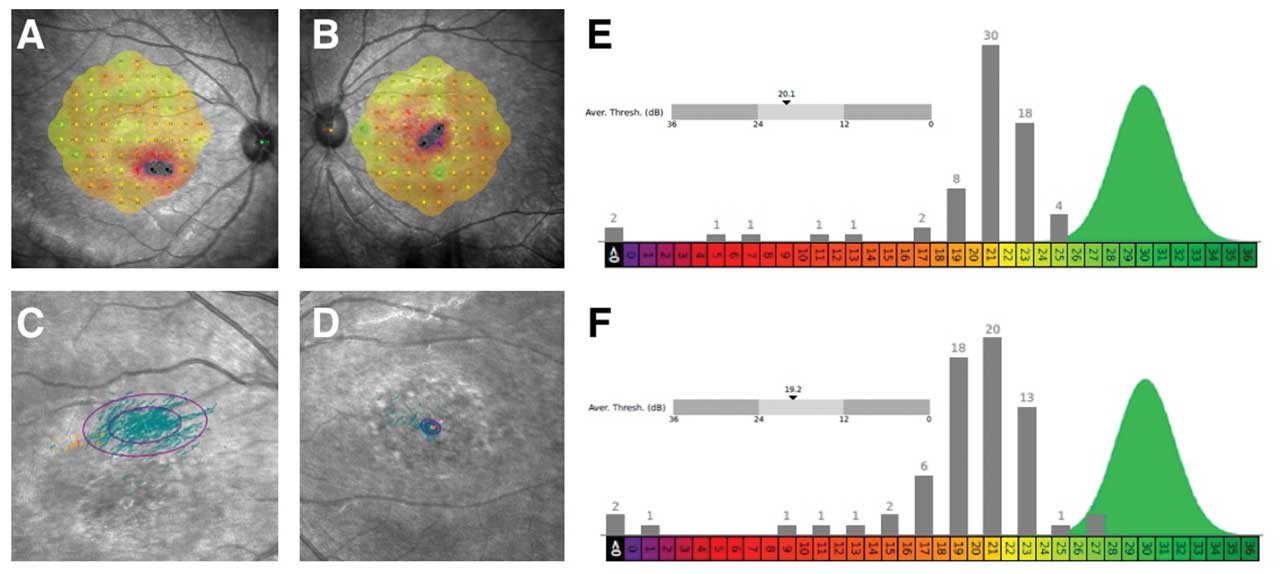
Figure 4. Microperimetry of a 20-year-old female patient with Stargardt disease indicates decreased central sensitivity of right eye (A) and left eye (B). Both eyes have no sensitivity (black) in 2 retinal loci near foveal center with surrounding moderate sensitivity loss (red). There is near normal sensitivities OU (green/yellow) in perifoveal retinal loci. There is a decentered preferred retinal locus in right eye (C) with large ellipse, consistent with unstable fixation. There is stable fixation in the left eye (D). The average threshold is 20.1 decibels (dB) and 19.2 dB in right eye (E) and left eye (F), respectively. The histograms of thresholds frequencies for both eyes are shown, showing abnormal sensitivities across most retinal loci in right eye (E) and left eye (F). This testing was performed on the same patient represented in Figure 1.
Finally, genetic testing is essential for confirming the diagnosis of Stargardt disease and identifying the specific variants in the ABCA4 gene. Next-generation sequencing and whole-exome sequencing are commonly used techniques with a detection rate of success around 70% to 80%.17 Inherited retinal dystrophy panels (targeted panels) exist that test for ABCA4 variants.
Navigating these new choices for genetic testing is complex for most busy retina surgeons. Importantly, there are unique ethical and practical implications of genetic testing, specifically including scope of genetic testing and pretest and posttest counseling. Further, results can return with variants of unknown significance (VUS) that legally require ongoing surveillance and interpretation, long after the visit. Finally, the costs, coverage, family implications, and phasing — determining with family testing if multiple mutations in the same gene are on one or different alleles of DNA to explain phenotype — all argue for genetic testing to be carried out by an experienced team with genetic counseling support.18
Other genes are known to cause Stargardt-like diseases that share similar phenotypes with Stargardt disease but are inherited in an autosomal dominant manner.19 These diseases include ELOVL4 and PROM1-related macular dystrophies. Both conditions can lead to retinal abnormalities that may resemble Stargardt disease. Additionally, variants in other genes, such as WDR19, PRPH2, CDH3, CERKL, CRX, BEST1, IMPG1, RDH12, and RPGR, can produce a similar phenotype, although they may lack the classic triad of flecks, macular atrophy, and peripapillary sparing seen in Stargardt disease.
Current Treatments and Management
Despite significant advancements in understanding the genetic and molecular basis of Stargardt disease, there is currently no cure for the condition. Treatment options are primarily focused on managing symptoms, slowing disease progression, and improving the quality of life for affected individuals.
One of the key management strategies for Stargardt disease is the avoidance of excessive vitamin A intake.20 Because vitamin A is a precursor of all-trans-retinal, reducing its intake may help decrease the accumulation of toxic byproducts in the retina. Patients are typically advised to avoid vitamin A supplements and foods high in vitamin A. This stands in contrast to retinitis pigmentosa, where vitamin A supplementation was traditionally thought to be beneficial, a long-held conclusion that recently was brought into question.21 As such, consultation with a dietician is recommended to help patients curate their diet. The goal is to eat vitamin A-rich foods in moderation with monitoring of daily retinol activity equivalents (RAE), a unit of measurement for vitamin A.20 Patients should not be asked to stop eating vitamin A-rich foods, as these foods constitute a healthy diet. In addition, some studies suggest that antioxidants and carotenoids, such as lutein and zeaxanthin, may provide protective benefits for the retina.22 These compounds are believed to reduce oxidative stress and protect photoreceptor cells from damage. However, their efficacy is still unclear to date.
As central vision deteriorates, patients with Stargardt disease often benefit from low-vision aids designed to enhance remaining vision. These aids include magnifying lenses, electronic magnifiers, screen readers, large print with high-contrast material, telescopes, and wearable artificial intelligence visual assistants. It is recommended that patients with Stargardt disease consult with an optometrist who has expertise in low-vision services; referrals to state services are also important as there is often need for improving home lighting, occupational therapy, and/or mobility training. Visual acuity, visual field testing, symptoms, and state-specific driving laws should be reviewed every year to ensure patients with driver’s licenses are safe to continue driving. Testing such as microperimetry can aid in determining the location of preferred retinal loci (PRL).
Emerging Treatments
Stargardt disease is currently the focus of extensive research and clinical development exploring various therapeutic avenues.23 Innovative strategies under investigation for Stargardt disease include pharmacologic treatments to slow disease progression. One promising regenerative medicine approach is gene therapy, which aims to introduce a functional copy of the ABCA4 gene into retinal cells to restore normal cellular function and prevent toxic buildup. Stem cell–based therapies are being explored to replace damaged retinal cells, potentially restoring vision. These diverse approaches highlight the dynamic and evolving landscape aimed at providing hope and improved quality of life for patients with Stargardt disease.
Observational Studies
There are currently 2 recruiting observational trials to study the progression of Stargardt disease. Patients aged 12 to 65 with macular lesions associated with biallelic Stargardt disease may be eligible for the Splice Bio trial.24 Another sponsor, Ascidian Therapeutics, is enrolling for an observational prescreening study for ACDN-01.25 These observational studies are imperative for choosing appropriate endpoints and outcomes and determining eligibility in interventional clinical trials.
Senolytic Agents
There is an investigational trial studying the effects of oral metformin on ABCA4-related retinopathy, including Stargardt disease.26 Metformin, a common diabetes medication, is known to have positive metabolic and lysosomal effects, specifically targeting RPE lysosomal and fatty acid oxidation pathways.27 This drug is being investigated for safety and efficacy at the National Eye Institute with recruitment of patients with Stargardt disease who are over age 12 and who have demonstrated a loss of vision over at least 2 years.
Slowing vitamin A dimers
Alkeus Pharmaceutical is investigating a new chemical entity, gildeuretinol (ALK-001), which inhibits the formation of toxic bisretinoids like A2E.28 A phase 1 clinical trial
confirmed the safety and tolerability of ALK-001, and a phase 2 trial showed it significantly slowed the progression of atrophic lesions in patients with late-stage Stargardt disease without major side effects, although no significant change in best-corrected visual acuity (BCVA) was detected.29 Another trial within the TEASE program is using sibling pairs to benchmark the younger siblings future retinal disease against the older siblings present condition.
Visual Cycle Modulators
Several pharmacologic strategies are being investigated to slow the progression of Stargardt disease by targeting different parts of the visual cycle. Emixustat hydrochloride (Kubota Vision) is an orally available visual cycle modulator that inhibits the RPE65 enzyme. This blockade in turn reduces lipofuscin production by slowing down the chemical process that transforms light into electrical signals in the retina, thereby blocking the accumulation of lipofuscin and vitamin A toxins. Phase 1 clinical trials showed it was well tolerated and slowed the rod visual cycle.30 In a phase 2 trial for Stargardt disease patients, emixustat demonstrated biologic activity by suppressing the visual cycle, particularly at a 10 mg dose.31 A phase 3 trial suggested that emixustat might slow disease progression in Stargardt disease patients with smaller lesions, showing a reduction in lesion progression compared to placebo over 24 months.32
Retinol-binding protein 4 (RBP4) antagonists are also being explored as potential therapies for Stargardt disease. RBP4 functions as a plasma transporter of retinol.33 Tinlarebant (Belite Bio), an RBP4 antagonist, is currently in a phase 3 trial to evaluate its ability to slow atrophic lesion growth in adolescent Stargardt patients.34 Similarly, STG-001 (Stargazer Pharmaceuticals) was shown in phase 1 clinical trials to reduce RBP4 levels.35 Further trials will be necessary to assess the success of this strategy.
Inflammatory Modulator
Avacincaptad pegol (Izervay; Astellas) is a complement factor C5 inhibitor administered via intravitreal injection; it has been approved by the FDA to treat geographic atrophy and is currently being investigated as a treatment for other retinal disorders, including Stargardt disease and age-related macular degeneration (AMD). Preclinical data suggest lipofuscin accumulation leads to complement activation and ultimately cell death.36 A phase 2/3 clinical trial for AMD demonstrated a significant reduction in geographic atrophy growth rates, though with increased macular neovascularization compared to sham controls.37 A phase 2b trial for Stargardt disease is ongoing.
Stem Cell Therapy
Stem cell–based therapies are another area of active research, with the potential to replace damaged RPE cells and restore vision in patients with Stargardt disease. The goal is to generate healthy RPE cells from stem cells and transplant them under the retina to replace the lost cells.
The first phase 1/2 human stem cell clinical trials for Stargardt disease in the United States were completed in 2015.38,39 These studies involved subretinal injections of human embryonic stem cell–derived retinal pigment epithelial cells in patients with late-stage Stargardt disease as well as atrophic AMD.40,41 Preliminary results showed no abnormal growth or immune rejection, with some improvement in visual acuity (VA) in treated eyes. The results from 9 Stargardt disease patients and 9 dry AMD patients demonstrated no serious adverse events related to the transplanted cells, though some adverse events were linked to the required immunosuppressive therapy. A third trial in the UK similarly showed no uncontrolled proliferation or inflammatory responses, but no significant changes in VA or quality of life at a 1-year follow-up.42
Gene Therapy
Gene therapy is a promising avenue of research for the treatment of Stargardt disease. The goal of gene therapy is to introduce a functional copy of the ABCA4 gene into the retinal cells, thereby restoring the normal transport of all-trans-retinal. Adeno-associated virus (AAV) is a gene therapy delivery system that has shown safety with minimal adverse events in trials, but its limited cargo capacity of 4.7 kb poses challenges for delivering large genes like ABCA4, which has a coding sequence of 6.8 kb.43 To overcome this, researchers have developed dual AAV vector strategies, such as fusing overlapping gene regions or using trans-splicing techniques to deliver full-length ABCA4 coding sequences.44 Preclinical studies have shown promising results, including successful expression of ABCA4 mRNA and full-length protein in knockout mice.45 Both Abeona Therapeutics and ViGeneron are working on this approach. However, gene therapy trials have been known to be complicated by progressive chorioretinal atrophy and persistent intraocular inflammation.46,47 Further modes of delivery may be necessary.
In a different approach, Ocugen’s OCU-410ST uses AAV5–human retinoic acid receptor–related orphan receptor alpha (hRORA) gene therapy to treat Stargardt disease as well as other inherited retinal diseases. As a gene-agnostic approach, this paradigm treats cellular dysfunction that may be applicable to multiple diseases. The results for hRORA show promise as a potential treatment for ABCA4-associated retinal diseases by improving retinal function and reducing toxic retinal byproducts in preliminary mouse studies.48 A phase 1/2 clinical trial is planned to further assess its safety and efficacy in humans.
Other strategies exist, such as optogenetics, that use AAV technology to deliver a light-sensitive ion channel to the inner retina of patients, which is not directly affected in Stargardt disease. The idea behind this technology is to generate a light-induced action potential through the retinal ganglion cell to the brain if the outer segments are already atrophic or missing in Stargardt disease or other inherited retinal diseases. Nanoscope Therapeutics conducted a phase 1/2a clinical trial in 2022 using an AAV vector to deliver a multicharacteristic opsin-I (vMCO-I) gene to bipolar cells.49 STARLIGHT is an ongoing phase 2 trial.50 Preliminary data presented to date suggest overall safety with potential visual improvement.51
ACDN-01 is a new technology by Ascidian Therapeutics being studied for Stargardt disease. The technology uses viral vector delivery of an RNA exon editor. While their results remain unpublished, STELLAR is an ongoing open-label phase 1/2 study to evaluate the safety and efficacy of a single dose of ACDN-01.52
Conclusion
Stargardt disease presents significant challenges due to its diversity of presentation, the progressive nature of the disease, and the current lack of curative treatments. However, the rapid advancements in innovative therapeutic approaches offer hope for the future. As researchers continue to unravel the molecular mechanisms underlying Stargardt disease and explore new treatment modalities, the prospects for preserving and even restoring vision for those affected by this debilitating condition are promising.
Ongoing research, clinical trials, and collaboration among scientists, clinicians, and patients will be essential in turning these possibilities into reality. The future of Stargardt disease treatment is bright, with the potential to transform the lives of countless individuals and bring new hope to those living with vision loss. RP


References
1. Walia S, Fishman GA. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 2009;30(2):63-68. doi:10.1080/13816810802695550
2. Stone EM, Andorf JL, Whitmore SS, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314-1331. doi:10.1016/j.ophtha.2017.04.008
3. Allikmets R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;17(1):122. doi:10.1038/ng0997-122a
4. Tsin A, Betts-Obregon B, Grigsby J. Visual cycle proteins: structure, function, and roles in human retinal disease. J Biol Chem. 2018;293(34):13016-13021. doi:10.1074/jbc.AW118.003228
5. Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67(4):960-966. doi:10.1086/303079
6. Parmar VM, Parmar T, Arai E, Perusek L, Maeda A. A2E-associated cell death and inflammation in retinal pigmented epithelial cells from human induced pluripotent stem cells. Stem Cell Res. 2018;27:95-104. doi:10.1016/j.scr.2018.01.014
7. Sheffield VC, Stone EM. Genomics and the eye. N Engl J Med. 2011;364(20):1932-1942. doi:10.1056/NEJMra1012354
8. Fishman GA, Farber M, Patel BS, Derlacki DJ. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. 1987;94(7):809-814. doi:10.1016/s0161-6420(87)33533-x
9. Strauss RW, Muñoz B, Wolfson Y, et al. Assessment of estimated retinal atrophy progression in Stargardt macular dystrophy using spectral-domain optical coherence tomography. Br J Ophthalmol. 2016;100(7):956-962. doi:10.1136/bjophthalmol-2015-307035
10. Lee W, Nõupuu K, Oll M, et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci. 2014;55(10):6139-6149. doi:10.1167/iovs.14-15126
11. Fujinami K, Lois N, Mukherjee R, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. 2013;54(13):8181-8190. doi:10.1167/iovs.13-12104
12. Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001;119(3):359-369. doi:10.1001/archopht.119.3.359
13. Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol. 2006;90(8):1060-1066. doi:10.1136/bjo.2006.097436
14. Cideciyan AV, Swider M, Aleman TS, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007;24(5):1457-1467. doi:10.1364/josaa.24.001457
15. Lad EM, Duncan JL, Liang W, et al. Baseline microperimetry and OCT in the RUSH2A study: structure-function association and correlation with disease severity. Am J Ophthalmol. 2022;244:98-116. doi:10.1016/j.ajo.2022.08.013
16. Foundation Fighting Blindness. FFB-CRI launching natural history study for people with USH2A mutations. April 4, 2017. Accessed September 30, 2024. https://www.fightingblindness.org/news/ffb-cri-launching-natural-history-study-for-people-with-ush2a-mutations-675
17. Stephenson KAJ, Zhu J, Dockery A, et al. Clinical and genetic re-evaluation of inherited retinal degeneration pedigrees following initial negative findings on panel-based next generation sequencing. Int J Mol Sci. 2022;23(2):995. doi:10.3390/ijms23020995
18. Jamal L, Schupmann W, Berkman BE. An ethical framework for genetic counseling in the genomic era. J Genet Couns. 2020;29(5):718-727. doi:10.1002/jgc4.1207
19. Kohli P, Tripathy K, Kaur K. Stargardt Disease. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2024. Updated January 8, 2024. Accessed September 9, 2024. https://www.ncbi.nlm.nih.gov/books/NBK587351/
20. Sofi F, Sodi A, Franco F, et al. Dietary profile of patients with Stargardt’s disease and retinitis pigmentosa: is there a role for a nutritional approach? BMC Ophthalmol. 2016;16:13. doi:10.1186/s12886-016-0187-3
21. Comander J, Weigel DiFranco C, Sanderson K, et al. Natural history of retinitis pigmentosa based on genotype, vitamin A/E supplementation, and an electroretinogram biomarker. JCI Insight. 2023;8(15):e167546. doi:10.1172/jci.insight.167546
22. Alsberge JB, Agarwal A. Late-onset Stargardt disease. Am J Ophthalmol Case Rep. 2022;26:101429. doi:10.1016/j.ajoc.2022.101429
23. Hussain RM, Ciulla TA, Berrocal AM, Gregori NZ, Flynn HW Jr, Lam BL. Stargardt macular dystrophy and evolving therapies. Expert Opin Biol Ther. 2018;18(10):1049-1059. doi:10.1080/14712598.2018.1513486
24. An observational study in subjects to follow the progression of Stargardt disease type 1 (STGD1) caused by bi-allelic autosomal recessive mutations in the ABCA4 gene (POLARIS). ClinicalTrials.gov identifier: NCT06435000. Updated September 26, 2024. Accessed October 1, 2024. https://clinicaltrials.gov/study/NCT06435000
25. Prescreening study to identify potential participants for ACDN-01 clinical trials. Clincialtrials.gov identifier: NCT06445322. Updated June 21, 2024. Accessed October 1, 2024. https://clinicaltrials.gov/study/NCT06445322
26. Oral metformin for treatment of ABCA4 retinopathy. ClinicalTrials.gov identifier: NCT04545736. Updated September 19, 2024. Accessed September 30, 2024. https://clinicaltrials.gov/study/NCT04545736
27. Bose DA, Farnoodian M, Sharma R, McGaughey D, Bharti K. Use of metformin as a potential treatment for Stargardt maculopathy. Presented at: Association for Research in Vision and Ophthalmology (ARVO) annual meeting; May 5-9, 2024; Seattle, Washington. https://iovs.arvojournals.org/article.aspx?articleid=2797549
28. Kaufman Y, Ma L, Washington I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem. 2011;286(10):7958-7965. doi:10.1074/jbc.M110.178640
29. Scholl HP. ALK-001 (C20-D3-vitamin A) slows the growth of atrophic lesions in ABCA4-related Stargardt disease: results of a phase 2 placebo-controlled clinical trial (TEASE study). Presented at: Association for Research in Vision and Ophthalmology (ARVO) annual meeting; May 1-4, 2022; Denver, Colorado.
30. Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, Birch D. Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina. 2012;32(1):183-188. doi:10.1097/IAE.0b013e318217369e
31. Kubota R, Birch DG, Gregory JK, Koester JM. Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. Br J Ophthalmol. 2022;106(3):403-408. doi:10.1136/bjophthalmol-2020-317712
32. Wang L, Shah SM, Mangwani-Mordani S, Gregori NZ. Updates on emerging interventions for autosomal recessive ABCA4-associated Stargardt disease. J Clin Med. 2023;12(19):6229. doi:10.3390/jcm12196229
33. Dobri N, Qin Q, Kong J, et al. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci. 2013;54(1):85-95. doi:10.1167/iovs.12-10050
34. Grigg JR, Chen F, Chen, TC, et al. Safety, tolerability, and efficacy of tinlarebant from the 24-month phase 2 study in adolescent patients affected by Stargardt disease. Presented at: Association for Research in Vision and Ophthalmology (ARVO) annual meeting; May 5-9, 2024; Seattle, Washington.
35. Study of STG-001 in subjects with Stargardt disease. ClinicalTrials.gov Identifier: NCT04489511. Updated April 27, 2021. Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT04489511
36. Radu RA, Hu J, Yuan Q, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286(21):18593-18601. doi:10.1074/jbc.M110.191866
37. Jaffe GJ, Westby K, Csaky KG, et al. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: a randomized pivotal phase 2/3 trial. Ophthalmology. 2021;128(4):576-586. doi:10.1016/j.ophtha.2020.08.027
38. Subretinal transplantation of hESC-derived RPE(MA09-hRPE) cells in patients with Stargardt’s macular dystrophy. ClinicalTrials.gov Identifier: NCT01345006. Updated July 7, 2021. Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT01345006
39. Safety and tolerability of sub-retinal transplantation of hESC-derived RPE (MA09-hRPE) cells in patients with advanced dry age-related macular degeneration (Dry AMD). ClinicalTrials.gov Identifier: NCT01344993. Updated July 7, 2021. Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT01344993
40. Schwartz SD, Hubschman JP, Heilwell G, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012;379(9817):713-720. doi:10.1016/S0140-6736(12)60028-2
41. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell–derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of 2 open-label phase 1/2 studies. Lancet. 2015;385(9967):509-516. doi:10.1016/S0140-6736(14)61376-3
42. Mehat MS, Sundaram V, Ripamonti C, et al. Transplantation of human embryonic stem cell–derived retinal pigment epithelial cells in macular degeneration. Ophthalmology. 2018;125(11):1765-1775. doi:10.1016/j.ophtha.2018.04.037
43. Grieger JC, Samulski RJ. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol. 2005;79(15):9933-9944. doi:10.1128/JVI.79.15.9933-9944.2005
44. McClements ME, MacLaren RE. Adeno-associated virus (AAV) dual vector strategies for gene therapy encoding large transgenes. Yale J Biol Med. 2017;90(4):611-623.
45. Dyka FM, Molday LL, Chiodo VA, Molday RS, Hauswirth WW. Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive Stargardt disease. Hum Gene Ther. 2019;30(11):1361-1370. doi:10.1089/hum.2019.132
46. Seitz IP, Wozar F, Ochakovski GA, et al. Dose-dependent progression of chorioretinal atrophy at the injection site after subretinal injection of rAAV2/8 in nonhuman primates. Ophthalmol Sci. 2024;4(5):100516. doi:10.1016/j.xops.2024.100516
47. Chan YK, Dick AD, Hall SM, et al. Inflammation in viral vector–mediated ocular gene therapy: a review and report from a workshop hosted by the Foundation Fighting Blindness, 9/2020. Transl Vis Sci Technol. 2021;10(4):3. doi:10.1167/tvst.10.4.3
48. Akula M, McNamee S, Chan NPM, DeAngelis MM, Haider NB. RORA modifier gene therapy rescues retinal degeneration in a juvenile AMD mouse model of Stargardt disease. Invest Ophthalmol Vis Sci. 2023;64(8):3842.
49. Dose-escalation study to evaluate the safety and tolerability of intravitreal vMCO-I in patients with advanced retinitis pigmentosa. ClinicalTrials.gov Identifier: NCT04919473. Updated June 9, 2021. Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT04919473
50. Safety and effects of a single intravitreal injection of vMCO-010 optogenetic therapy in subjects with Stargardt disease (STARLIGHT). ClinicalTrials.gov Identifier: NCT05417126. Updated November 9, 2023 . Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT05417126
51. Mahajan VB. Longitudinal BCVA analysis of patients with stargardt disease and macular degeneration treated with MCO-010, a mutation-agnostic optogenetic therapy: 48-week results from a phase 2a clinical trial (STARLIGHT). Presented at: Association for Research in Vision and Opthalmology (ARVO) annual meeting; May 5-9, 2024; Seattle, WA. Accessed September 30, 2024. https://iovs.arvojournals.org/article.aspx?articleid=2795392
52. Study to Evaluate ACDN-01 in ABCA4-related Retinopathy (STELLAR). ClinicalTrials.gov Identifier: NCT06467344. Updated June 25, 2024. Accessed September 9, 2024. https://clinicaltrials.gov/study/NCT06467344