







Uveitis is a group of inflammatory intraocular diseases affecting the uvea. It is one of the leading causes of preventable vision loss in the United States and continues to be a diagnostic conundrum, in part due to the heterogeneity of disease presentation. In any patient with suspected uveitis, infectious etiologies should always be considered and treated first. However, most uveitis is noninfectious in etiology and many of these cases are either idiopathic (37%)1 or associated with a diverse group of autoimmune diseases, including sarcoidosis, Behcet disease, Vogt-Koyanagi-Harada (VKH) disease, and tubulointerstitial nephritis and uveitis (TINU) syndrome. Left untreated, some of these conditions can be life threatening. Patients with these diseases may present initially to the vitreoretinal specialist with ocular involvement as the first manifestation of their systemic autoimmune disease. Therefore, familiarity with a methodical evaluation of the uveitis patient and maintaining a high degree of suspicion for autoimmune-associated disease will maximize both ocular and systemic outcomes for these challenging and often medically complex cases.
GENERAL PRINCIPLES OF UVEITIS EVALUATION
Diagnostic workup is guided by a thorough history-taking and ophthalmologic exam. Often, a patient will be referred for multidisciplinary management of a known autoimmune condition. However, some patients may present with ocular symptoms of a yet-undiagnosed systemic disease. Performing a comprehensive review of systems helps screen for other organ systems affected by a unifying diagnosis.
Uveitis is classified based on the affected anatomic location (anterior, intermediate, posterior, or panuveitis). A careful ophthalmologic exam detailing the ocular structures affected, laterality, presence or absence of granulomatous inflammation, and intraocular pressure (IOP) abnormalities is crucial to narrowing a differential diagnosis for the patient. In patients with evidence of posterior-segment inflammation, multimodal imaging may provide additional evidence of active inflammation that may not be visible on exam. Optical coherence tomography (OCT) of the macula can screen for the presence of cystoid macular edema (CME), a frequent complication of uveitis. Fluorescein angiography (FA) can reveal papillitis and vasculitis, suggesting active disease, that may not be apparent on exam. Indocyanine green (ICG) angiography can be considered in patients with suspected choroidal involvement and can help distinguish posterior segment–involving uveitis from other entities such as central serous chorioretinopathy.
After obtaining an appropriate history, exam, and imaging, the next step is to rule out syphilis — a common cause of uveitis — and tuberculosis, which is less common in the United States yet endemic in other areas of the world. Testing for tuberculosis is important not only because it may be the etiology of uveitis, depending on the ocular features of the disease, but because it may affect management. High-dose systemic corticosteroid or other immunosuppression is often used in the treatment of noninfectious uveitis, and even if the uveitis is not tuberculosis related, it is important to determine if the patient may have latent tuberculosis in addition to noninfectious uveitis, because immunosuppressive therapies may lead to reactivation of tuberculosis.
If IOP is elevated and there is suspicion for a herpesvirus, ocular fluid sampling and polymerase chain reaction testing should be considered. Subsequent workup is informed by a differential diagnosis of possible autoimmune diseases, which can be guided by screening for concurrent systemic symptoms. Below is a review of systemic diseases with associated uveitis frequently seen in the retina clinic.
SARCOIDOSIS
Sarcoidosis is a systemic noncaseating granulomatous autoimmune disease that most commonly affects the lungs, skin, lymph nodes, and eyes. While granulomatous anterior uveitis is the most common manifestation of sarcoidosis in the eye, ocular sarcoid (OS) is not an exclusively granulomatous disease. Ocular sarcoid is classified based on intraocular and systemic signs into definite, presumed, and probable disease (Table 1).2 Definite OS is defined as biopsy-proven with compatible uveitis. A patient with bilateral hilar adenopathy and at least 2 intraocular signs, but without a confirmatory biopsy, has presumed OS. Probable OS patients have neither a positive biopsy nor bilateral hilar adenopathy but do have at least 3 intraocular and 2 systemic signs of sarcoidosis. Ocular sarcoid–related posterior uveitis commonly presents with macular edema and multifocal chorioretinitis that can be associated with or without vasculitis (Figure 1). Choroidal granulomas are often suggestive of OS but are typically asymptomatic unless they are peripapillary or involve the macula.3 Fluorescein angiography, ICG angiography, and OCT are all useful tools in monitoring posterior-segment disease activity in these patients.
| OTHER CAUSES OF GRANULOMATOUS UVEITIS ARE RULED OUT | |
|---|---|
| INTRAOCULAR SIGNS | SYSTEMIC SIGNS |
| Mutton fat keratic precipitates and/or iris nodules (Koeppe, Busacca) | Bilateral hilar lymphadenopathy |
| Trabecular meshwork nodules | Negative tuberculin test or interferon-gamma releasing assay (Quantiferon gold) |
| Vitreous opacities (snowballs or string of pearls) | Elevated serum angiotensin-converting enzyme |
| Multiple peripheral chorioretinal lesions | Elevated serum lysozyme |
| Nodular or segmental periphlebitis or macroaneurysms | Elevated CD4/CD8 ratio (>3.5) in bronchoalveolar lavage fluid |
| Optic disc or choroidal granulomas | Abnormal positron emission tomography imaging |
| Bilaterality of presentation | Lymphopenia |
| Parenchymal lung disease consistent with sarcoid | |
BEHCET DISEASE
Behcet disease is a systemic inflammatory disease that frequently affects the eyes with a relapsing and remitting nongranulomatous panuveitis with retinal vasculitis. It is strongly associated with the major histocompatibility complex antigen HLA-B51 and classically presents as a “triple-symptom complex” of recurrent aphthous oral ulcers, genital ulcers, and uveitis. Behcet disease is a clinical diagnosis based on criteria developed by the International Study Group for Behcet disease (Table 2).
| DIAGNOSTIC CRITERIA | ADDITIONAL SYMPTOMS |
|---|---|
| Recurrent oral aphthous ulcers (at least 3 episodes in 1 year) with 2 of the additional symptoms from column 2. | Recurrent genital ulcers |
| Cutaneous lesions: erythema nodosum, pseudovasculitis, papulopustular lesions, acneiform lesions | |
| Ocular lesions: anterior, intermediate, or posterior uveitis; retinal vasculitis | |
| Positive pathergy test: papule >2 mm after 24 to 48 hours of antigen inoculation |
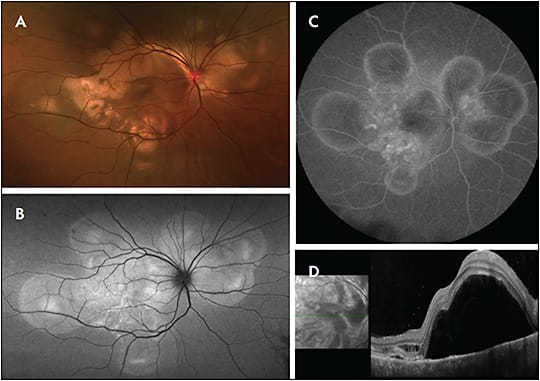
Behcet uveitis can often present with a “cold” hypopyon, or without ciliary flush.4 This contrasts with the “hot” hypopyon seen in HLA-B27–associated uveitis. Posterior-segment inflammation, a vision-threatening manifestation of Behcet disease, is characterized by vitritis and retinal vasculitis that can affect both arterioles and venules (Figure 2). Transient retinal infiltrates can also be seen. As the remitting course of disease causes cumulative ischemic damage, patients can develop postinflammatory complications, such as retinal atrophy and neovascular glaucoma.5 Immunomodulatory therapy, specifically tumor necrosis factor inhibitors, should be strongly considered in patients with Behcet disease with uveitis.6
VOGT-KOYANAGI-HARADA DISEASE
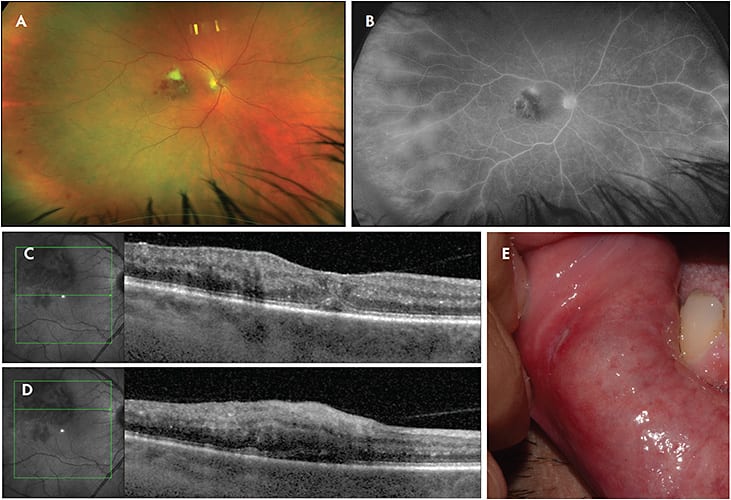
Vogt-Koyanagi-Harada disease (VKH) is a systemic granulomatous disease affecting the eyes, meninges, and skin. The mechanism of disease is suggested to be an autoimmune process directed at antigens associated with melanocytes. It most commonly affects peoples of Hispanic, Asian, Native American, or Middle Eastern descent. It presents as a spectrum of disease, for which the diagnostic criteria are categorized into complete, incomplete, and probable.7 In its early stages, VKH typically presents with a diffuse choroiditis, which can manifest from focal areas of subretinal fluid to larger bullous detachments (Figure 3A). Hyperautofluorescence is typically seen in the areas of subretinal fluid (Figure 3B). Patchy hyperfluorescence with pinpoint leakage is seen on fluorescein angiography during early VKH (Figure 3C). Ocular depigmentation, classically seen as pale choroidal depigmentation (sunset glow fundus) or perilimbal vitiligo (Sugiura sign), is a notable characteristic of the late stages of VKH. These ocular findings, which are bilateral, are often associated with neurologic (meningismus, tinnitus, cerebrospinal fluid pleocytosis) and integumentary (alopecia, vitiligo) findings in complete VKH disease. Incomplete VKH is defined as ocular symptoms with only 1 sign of neurologic or integumentary involvement, whereas probable VKH presents with no extraocular manifestations. For patients diagnosed with incomplete or probable VKH, additional systemic involvement may be noted later in the disease course.
TUBULOINTERSTITIAL NEPHRITIS AND UVEITIS SYNDROME
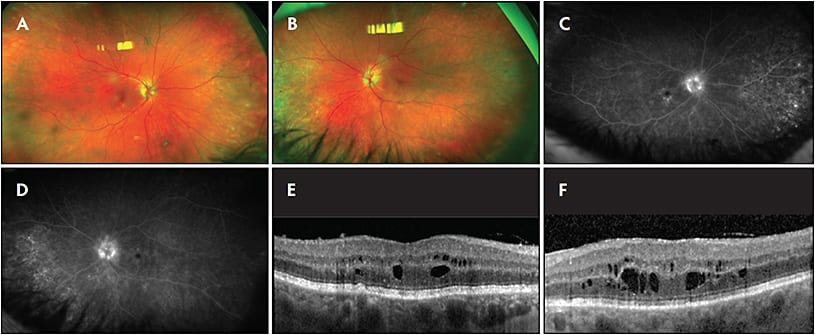
Tubulointerstitial nephritis and uveitis syndrome (TINU) is a rare autoimmune disease that presents with acute interstitial nephritis and uveitis without other organ involvement. TINU classically affects young females and most commonly presents in the eye as a bilateral nongranulomatous anterior uveitis. Posterior-segment involvement is increasingly recognized, particularly with the use of multimodal imaging. Interstitial nephritis is diagnosed by biopsy or by clinical criteria. “Complete” clinical criteria are met when the patient presents with elevated serum creatinine; abnormal urinalysis (Table 3); and systemic symptoms for a duration of 2 or more weeks consisting of any combination of fever, fatigue, or weight loss. The patient will also have and laboratory abnormalities, such as anemia, abnormal liver function, eosinophilia, or elevated erythrocyte sedimentation rate. Incomplete clinical criteria are met when fewer than 3 of the above are present. Definite TINU is defined as bilateral nongranulomatous anterior uveitis with or without intermediate or posterior segment involvement diagnosed less than 2 months before, or less than 12 months after, interstitial nephritis, and interstitial nephritis diagnosed either by biopsy or complete clinical criteria. Patients presenting with uveitis and incomplete interstitial nephritis clinical criteria have probable TINU. Patients who present with atypical uveitis and incomplete interstitial nephritis clinical criteria have possible TINU. On imaging, posterior involvement presents heterogeneously. While CME and vascular leakage are commonly seen, some eyes can have an unremarkable OCT with peripheral leakage on FA.8 Therefore, in patients with suspected TINU, fluorescein angiography should be considered even when the corresponding OCT is unremarkable.
| Increased urinary beta-2 microglobulin |
| Proteinuria |
| Urinary eosinophils |
| Pyuria |
| White cast cells |
| Normoglycemic glucosuria |
TREATMENT
Successful treatment of noninfectious uveitis associated with systemic autoimmune disease requires a multidisciplinary approach. Depending on the organs involved, management may involve collaboration between the ophthalmologist and various other subspecialists, including the rheumatologist, neurologist, pulmonologist, and nephrologist. Often, high-dose systemic corticosteroid, such as oral prednisone or intravenous methylprednisolone, and/or local steroid injections are needed to achieve quick control of ocular involvement. Systemic autoimmune disease frequently requires chronic treatment with immunomodulatory therapy, and an appropriate agent should be selected that can treat the involved organs, including the eyes. If the uveitis remains active despite attempted treatment with immunomodulatory therapy, treatment with long-acting local steroid formulations, such as the surgically implanted fluocinolone acetonide implant (Retisert; Bausch + Lomb), as well as the injectable fluocinolone acetonide implant (Yutiq; Eyepoint Pharmaceuticals), should be considered. Recurrent episodes of breakthrough inflammation may be controlled locally as well by topical steroid therapy, posterior-sub Tenon’s triamcinolone (Kenalog; Bristol-Myers Squibb) injection, suprachoroidal triamcinolone acetonide (Xipere; Bausch + Lomb) injection, or the injectable dexamethasone implant (Ozurdex; Allergan). Close attention should be paid to the risk of cataract formation and IOP elevation with use of local steroid treatment.
SUMMARY
Uveitis is a vision-threatening disease that requires careful evaluation. While it is necessary to consider infectious etiologies, most uveitis is noninfectious, and the vitreoretinal specialist should have a high suspicion for the above-mentioned autoimmune systemic diseases when these patients present to the retina clinic. Left untreated, uveitis can lead to irreversible profound vision loss, but fortunately for most patients today, excellent outcomes can be achieved with appropriate treatment.
Because most patients with systemic autoimmune disease with associated uveitis will require chronic treatment, a multidisciplinary approach is necessary to select immunomodulatory therapy that will treat the involved organ systems, including the eye. Sometimes, despite immunomodulatory therapy, uveitis may remain active, in which case local steroid may be necessary to achieve quiescence, with consideration given to the risk of cataract formation and IOP elevation. While challenging, the evaluation and management of these complex cases provides an exciting opportunity for the vitreoretinal specialist to play a significant part in improving patients’ quality of life and assist in the diagnosis and treatment of a potentially vision-threatening and life-threatening condition. RP
REFERENCES
- Choi RY, Rivera-Grana E, Rosenbaum JT. Reclassifying idiopathic uveitis: lessons from a tertiary uveitis center. Am J Ophthalmol. 2019;198:193-199. doi:10.1016/j.ajo.2018.10.018
- Mochizuki M, Smith JR, Takase H, et al. Revised criteria of International Workshop on Ocular Sarcoidosis (IWOS) for the diagnosis of ocular sarcoidosis. Br J Ophthalmol. 2019;103(10):1418-1422. doi:10.1136/bjophthalmol-2018-313356
- Matsou A, Tsaousis KT. Management of chronic ocular sarcoidosis: challenges and solutions. Clin Ophthalmol. 2018;12:519-532. doi:10.2147/OPTH.S128949
- Tugal-Tutkun I. Behçet’s Uveitis. Middle East Afr J Ophthalmol. 2009;16(4):219-224. doi:10.4103/0974-9233.58425
- Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgancioglu M. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373-380. doi:10.1016/j.ajo.2004.03.022
- Levy-Clarke G, Jabs DA, Read RW, Rosenbaum JT, Vitale A, Van Gelder RN. Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology. 2014;121(3):785-96.e3. doi:10.1016/j.ophtha.2013.09.048
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification criteria for Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2021;228:205-211. doi:10.1016/j.ajo.2021.03.036
- Cao JL, Srivastava SK, Venkat A, Lowder CY, Sharma S. Ultra-widefield fluorescein angiography and OCT findings in tubulointerstitial nephritis and uveitis syndrome. Ophthalmol Retina. 2020;4(2):189-197. doi:10.1016/j.oret.2019.08.012