







Sickle cell disease (SCD) is the most common inherited hematologic disorder and affects approximately 3.2 million people worldwide.1 It is most prevalent in populations of African descent, followed by those of Mediterranean, Caribbean, and South and Central American descent.2 Patients with SCD most commonly present with painful vaso-occlusive crises (VOC), particularly under hypoxic conditions. Other systemic complications, including acute chest syndrome, pulmonary hypertension, splenic sequestration, and cerebrovascular accidents, contribute to the significant morbidity and mortality of SCD.2
Sickle cell disease encompasses a group of hemoglobinopathies characterized by a mutation in the β-globin gene, which results in defective oxygen transport. Under hypoxic and inflammatory conditions, hemoglobin polymerization can result in sickling, or the formation of rigid and elongated erythrocytes, which subsequently cause intravascular hemolysis and thrombosis.3 Additional mechanisms, including endothelial dysfunction, vaso-occlusion, and inflammation, are thought to be involved in the pathophysiology of SCD.4
GENETICS
Adult hemoglobin (Hb A) consists of 2 α-globin and 2 β-globin subunits combined with a central heme. A glutamine-to-valine mutation in the sixth amino acid position of β-globin results in hemoglobin S (Hb S) whereas a glutamine-to-lysine mutation produces hemoglobin C (Hb C). Several combinations of β-globin subunits can be inherited and can produce homozygous SCD (HbSS) or the compound heterozygous SCD (HbSC).5 Other compound heterozygous forms also exist with less common mutations in β-globin. Homozygous SCD results in more severe systemic morbidity and mortality compared to HbSC disease.6 However, retinal involvement of SCD is more frequently associated with HbSC disease than HbSS disease.7 Patients with 1 normal and 1 abnormal β-globin gene (HbAS) have sickle cell trait and are largely asymptomatic, with the exception of sight-threatening ocular hypertension in the presence of hyphema, or in instances of severe hypoxia and dehydration.8 Sickle cell trait is rarely associated with retinal involvement.
SICKLE CELL RETINOPATHY
Retinal findings in SCD can be divided into nonproliferative sickle retinopathy and proliferative sickle retinopathy (PSR) when pathologic neovascularization develops. The main nonproliferative findings include salmon patch hemorrhages, iridescent spots, and black sunbursts. Salmon patch hemorrhages represent areas of “blowout” of an occluded vessel, whereas black sunbursts are hyperpigmented areas caused by intraretinal hemorrhage and are often associated with retinal pigment epithelium damage.9 Iridescent spots represent hemosiderin-laden macrophages in areas of prior intraretinal hemorrhage.10
Vaso-occlusion of peripheral retinal arterioles, progressive vascular remodeling, and retinal ischemia may result in PSR. In a longitudinal study, by age 24 to 26 years, 43% of people with HbSC disease were noted to develop PSR compared to 14% of those with HbSS disease.11 It remains unclear why HbSC more frequently leads to PSR compared to HbSS disease. However, several theories have been posited, including differences in cellular rheology and fetal hemoglobin content, as well as cellular responses to acute and chronic hypoxia.12,13 Patients with HbSC have a higher hematocrit, and thus more abnormal, sickled cells remain in circulation with delayed transit time through the retinal microvasculature.12,14 Decreased oxygenation results in indolent release of growth factors such as vascular endothelial growth factor (VEGF) and others.15 In HbSS disease, hemoglobin values are likely lower than those noted in HbSC. More complete vaso-occlusions may occur, with less likely release of proangiogenic growth factors.
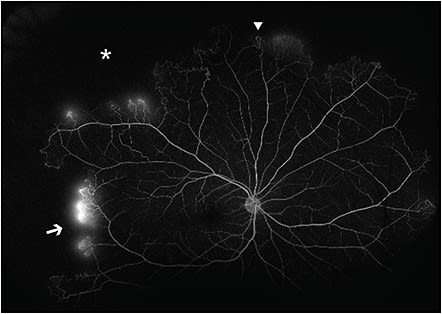
The Goldberg classification for PSR consists of 5 stages.12 Stage I PSR shows peripheral vascular occlusion, which can manifest as silver-wiring of the arterioles affected.16 The subsequent vascular remodeling at the border of perfused areas can lead to arteriovenous (AV) anastomoses, which are noted in stage II PSR. Stage III PSR consists of neovascular and fibrous proliferations. This includes the hallmark sea-fan neovascularization associated with sickle cell retinopathy (SCR). These are often found superotemporally at the border of perfused and nonperfused areas and are thought to arise from the venous component of AV anastomoses.12 Unlike AV anastomoses, sea-fan neovascularization demonstrates leakage on fluorescein angiography (Figure 1). Stage IV PSR is defined by the presence of vitreous hemorrhage caused by traction on neovascular fronds. Lastly, stage V PSR consists of tractional or combined rhegmatogenous and tractional retinal detachments. These are caused by vitreoretinal traction from neovascularization on ischemic retina, which is pathologically thin and thus more susceptible to retinal breaks. These detachments most commonly involve the ischemic temporal retinal periphery, a watershed zone for retinal vascular perfusion.12
SICKLE CELL MACULOPATHY
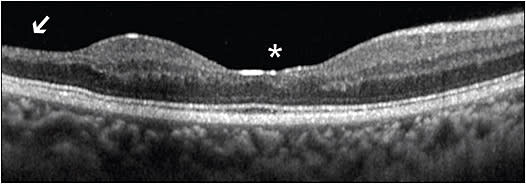
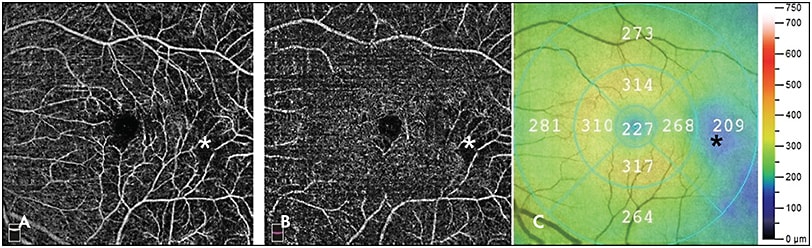
Although peripheral vascular changes are the hallmark features of SCR, maculopathy has also been detected in 45.6% of patients with SCD, with approximately equal prevalence in HbSS and HbSC disease.17,18 Enlarged foveal avascular zone and macular thinning due to ischemia, as well as foveal “splaying” or blunting of the foveal contour, are observed in sickle cell maculopathy.19-21 Temporal macular thinning has also been noted in SCD and is due to involvement of the watershed zone near the horizontal raphe (Figure 2).22 This watershed zone in the temporal macula, retinal field 3, may be analogous to the terminal circulation of the peripheral temporal retina.23 Moreover, nonperfusion of the macular deep capillary plexus on optical coherence tomography angiography (OCTA) has been associated with other areas of macular thinning (Figure 3A-3C).24 Interestingly, in a large case series of children with SCD, vascular changes were shown to precede structural changes in retinal thickness, further supporting a causative relationship.25
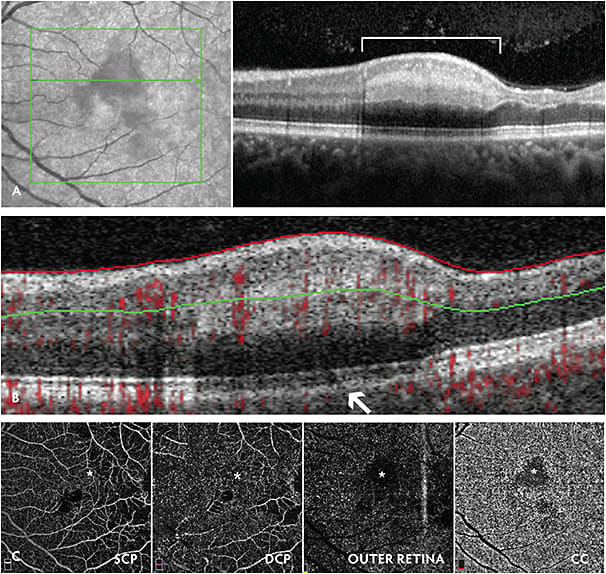
Patients with SCD can present with sudden vision loss due to retinal arteriolar occlusions, central retinal artery occlusion (rarely), or acute infarction of the macular choroidal circulation.26-29 In the latter cases, acute macular neuroretinopathy, or paracentral acute middle maculopathy can be observed, which are well visualized on spectral-domain optical coherence tomography (SD-OCT; Figure 4A-4C).30-32 These macular infarcts are most commonly observed in patients with HbSS disease. Although these patients can present with significant central scotomas, fortunately, vision loss is often self-limited. We advocate for urgent hematology consultation for consideration of systemic SCD assessment, including hydration, oxygenation, and red blood cell exchange transfusion for SCD patients who present with new retinal vascular occlusions or infarctions.
VISION LOSS IN SICKLE CELL DISEASE
Vision loss in SCD most often results from advanced stages of PSR.12,33 The most common causes of severe vision loss (visual acuity of 20/200 or worse) are vitreous hemorrhage and retinal detachment, which account for 44% and 31% of cases of severe vision loss, respectively.33 Less common causes include epiretinal membrane and macular hole. Overall, decreased visual acuity occurs in 10% to 12% of eyes with untreated PSR.33 Interestingly, foveal avascular zone changes have not been associated with a change in visual acuity, whereas temporal macular atrophy has been shown to correlate with scotomas and decreased contrast sensitivity.20,22,34
MEDICAL MANAGEMENT
The main goal of treatment is to prevent progression to Goldberg stage IV and stage V PSR, which are associated with vision loss. Proliferative lesions have been shown to autoinfarct and spontaneously regress in 32% of eyes.11 Therefore, intervention may not be indicated for small asymptomatic peripheral sea-fan neovascular lesions and lesions that appear to have autoinfarcted with decreased vascularization. However, treatment may be required for rapidly growing and vascularized proliferative lesions. Scatter laser photocoagulation of ischemic areas is the mainstay of treatment for PSR. Laser-treated patients with PSR showed a slightly decreased risk of vision loss from vitreous hemorrhage compared to those with PSR managed with observation.35 Laser has not been shown to decrease incidence of new sea-fan neovascularization. The proangiogenic growth factors hypoxia-inducible factor 1α (HIF-1α) and VEGF are highly expressed in ischemic areas in eyes with PSR, particularly at the boundary of perfused and nonperfused retina. Laser photocoagulation may suppress secretion of these growth factors. We advocate for application of scatter laser not only to areas of peripheral ischemic retina, but also to the marginal zone between ischemic and nonischemic retina, as well as just posterior to the marginal zone due to areas of growth factor expression.36
Intravitreal anti-VEGF agents have also been shown to result in partial to complete regression of retinal neovascularization.37-40 Off-label use of intravitreal bevacizumab (Avastin; Genentech) has been described as a helpful adjunctive treatment in PSR to facilitate resolution of vitreous hemorrhage in cases where the posterior segment view does not permit application of scatter laser, or when scatter laser has been applied and sea-fan neovascularization remains vascularized despite adequate prior laser treatment.37-39 Currently, we treat with intravitreal bevacizumab based on sea-fan vascularization activity determined by clinical exam and fluorescein angiography leakage. Preoperative intravitreal bevacizumab and sea-fan regression has also facilitated surgical management of retinal detachment in PSR in a case report.41 Of note, intravitreal anti-VEGF injections should be used with caution in patients with proliferative retinopathies and vitreous hemorrhage in the absence of a posterior vitreous detachment, because anti-VEGF therapy may accelerate fibrosis and contraction of peripheral neovascularization. Rapid contraction of the sea-fan neovascular complex may cause increased traction on the underlying retina, and thus may increase the risk of a resultant retinal detachment.42 Larger studies investigating the role of anti-VEGF therapies in PSR treatment are needed to inform best practices and optimal treatment regimens.
SURGICAL MANAGEMENT
Surgical management is reserved for nonclearing vitreous hemorrhage, bilateral vitreous hemorrhage, or vitreous hemorrhage in a monocular patient, as well as retinal detachment. Scleral buckling in sickle cell retinal detachment was historically associated with higher rates of anterior-segment ischemia. However, the incidence of anterior-segment ischemia has declined significantly with modern surgical techniques. Surgeons should avoid broad and/or scleral buckling elements, compression of the long posterior ciliary vessels, excessive cryopexy, and extraocular muscle injury in patients with SCD.43,44
Vitrectomy has become a common surgical technique for the management of PSR-associated retinal detachments. The segmentation technique for eliminating fibrovascular traction is preferred to minimize traction on the ischemic peripheral retina and to avoid iatrogenic retinal breaks. This technique involves vertical cuts through the fibrovascular tissue. Delamination, in which fibrovascular membranes are horizontally dissected from the retina to relieve tangential retinal traction, may increase likelihood of iatrogenic retinal breaks.45 Due to the thin ischemic peripheral retina in eyes with PSR, repair of retinal detachments is more frequently associated with iatrogenic tears and lower rates of single surgery success.45 We advocate for bimanual vitreoretinal surgical techniques when possible. Additional careful placement of encircling or segmental scleral buckling can be used to support peripheral pathology, avoiding high encircling elements.
Careful preoperative planning with the anesthesiology and hematology teams is recommended to optimize surgical outcomes for SCD patients undergoing vitreoretinal surgery.46 This includes adequate hydration and supplemental oxygen intraoperatively and postoperatively, as well as a warm operating room. General anesthesia or local anesthesia with a sub-Tenon’s block are preferred over retrobulbar block to avoid elevated intraocular pressure and occlusion of the central retinal artery. During surgery, it is also essential to maintain careful control of intraocular pressure to avoid prolonged retinal artery occlusion.8 Postoperative monitoring of hydration, oxygenation, analgesia, and intraocular pressure is also crucial. Routine perioperative transfusion may be considered on an individualized basis in consultation with a hematologist.44,46
SCREENING AND MONITORING
Newborn sickle cell screening has been mandated in all 50 states in the United States since 2006.47 However, patients may be unaware of their sickle cell status or may not have been screened if they were born elsewhere or before the newborn screening implementation. Therefore, patients may have SCD or other undiagnosed hemoglobinopathy. It is reasonable to obtain a serum electrophoresis on patients with ocular pathology suspicious for SCR, even with a negative past medical history of SCD. Currently, there are no evidence-based screening guidelines for SCR. Expert consensus recommendations advise referral for patients with SCD for retinal exam yearly beginning at age 10, with annual evaluation thereafter.48 Several imaging modalities are used to assess and monitor peripheral PSR, including ultrawidefield imaging and fluorescein angiography.49 In addition, SD-OCT and OCTA may be used to monitor structural and vascular alterations in the macula.
In a recent retrospective cohort study on pediatric SCR, the average age of onset of PSR was 12.2 years in patients with HbSC disease and 15.4 years in those with HbSS disease. Based on these findings, these authors recommend screening for treatment-requiring PSR at age 9 and age 13 for asymptomatic patients with HbSC and HbSS, respectively.50 Further research may inform retina specialists’ practice patterns in SCR surveillance exams and help establish screening guidelines. Moreover, there is ongoing research on the use of teleophthalmology with ultrawidefield fundus imaging, as well as machine learning techniques to detect and classify the stage of SCR.51 This technology may play a role in SCR screening and monitoring in the future.
SYSTEMIC MANAGEMENT
Given the recent advances in medical therapy for SCD, a brief discussion on the systemic management of SCD is warranted. Allogeneic stem-cell therapy from human leukocyte antigen-matched donors is the only known cure for SCD, although gene therapy has shown promise in clinical trials to date.52,53 Blood transfusions are also used long-term to decrease the Hb S content of blood. For many years, hydroxyurea, which increases fetal hemoglobin content, was the only approved oral therapy for SCD.54 In 2019, 2 new oral therapies were approved for SCD. Voxelotor (Oxbryta; Global Blood Therapeutics), which inhibits Hb S polymerization, and crizanlizumab (Adakveo; Novartis), a monoclonal antibody that binds P-selectin, were shown to reduce hemolysis and prevent VOC respectively.55,56 Longitudinal studies involving patients receiving these novel therapies will help understand the effect of these therapies on SCR.
CONCLUSION
Although most patients with SCD generally have good distance vision, SCR can cause significant visual morbidity most commonly from vitreous hemorrhage and retinal detachment. Patients with SCD can also present with acute vision loss from retinal vascular occlusions or macular infarctions. Laser photocoagulation is the mainstay treatment for PSR, and intravitreal anti-VEGF therapy has also been effective as an adjunctive agent in select cases of PSR. Retinal detachment related to PSR can be challenging to manage surgically. Therefore, these authors recommend all patients with SCD receive regular exams with a retina specialist at least annually for SCR surveillance, with consideration of ultrawidefield fundus photography and fluorescein angiography, and with macular OCT, to document baseline retinal anatomic changes and to monitor for future disease progression. Additional study is necessary to understand the relationship between systemic SCD therapy, retinal perfusion status, and incidence and severity of PSR. RP
REFERENCES
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117-171.
- Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55(4):359-377.
- Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. 2010;12:345-367.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019;14:263-292.
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151(9):839-845.
- Ballas SK, Lewis CN, Noone AM, Krasnow SH, Kamarulzaman E, Burka ER. Clinical, hematological, and biochemical features of Hb SC disease. Am J Hematol. 1982;13(1):37-51.
- Lutty GA GM. Ophthalmological complications. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, eds. Sickle cell disease: basic principles and clinical practice. New York, NY: Raven Press; 1994:703-724.
- Goldberg MF. The diagnosis and treatment of secondary glaucoma after hyphema in sickle cell patients. Am J Ophthalmol. 1979;87(1):43-49.
- Gagliano DA, Goldberg MF. The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol. 1989;107(12):1814-1815.
- Romayanada N, Goldberg MF, Green WR. Histopathology of sickle cell retinopathy. Trans Am Acad Ophthalmol Otolaryngol. 1973;77(5):OP652-OP676.
- Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112(11):1869-1875.
- Goldberg MF. Retinal neovascularization in sickle cell retinopathy. Trans Am Acad Ophthalmol Otolaryngol. 1977;83(3 Pt 1):Op409-431.
- Ansari J, Gavins FNE. Ischemia-reperfusion injury in sickle cell disease: from basics to therapeutics. Am J Pathol. 2019;189(4):706-718.
- Elagouz M, Jyothi S, Gupta B, Sivaprasad S. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55(4):359-377.
- Kurihara T, Westenskow PD, Friedlander M. Hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF) signaling in the retina. Adv Exp Med Biol. 2014;801:275-281.
- Nagpal KC, Goldberg MF, Rabb MF. Ocular manifestations of sickle hemoglobinopathies. Surv Ophthalmol. 1977;21(5):391-411.
- Beral L, Romana M, Lemonne N, et al. Multifocal electroretinogram findings in sickle cell maculopathy. Eye (Lond). 2019;33(12):1939-1945.
- Leitao Guerra RL, Leitao Guerra CL, Bastos MG, de Oliveira AHP, Salles C. Sickle cell retinopathy: What we now understand using optical coherence tomography angiography. A systematic review. Blood reviews. 2019;35:32-42.
- Goldbaum MH. Retinal depression sign indicating a small retinal infarct. Am J Ophthalmol. 1978;86(1):45-55.
- Sanders RJ, Brown GC, Rosenstein RB, Magargal L. Foveal avascular zone diameter and sickle cell disease. Arch Ophthalmol. 1991;109(6):812-815.
- Hoang QV, Chau FY, Shahidi M, Lim JI. Central macular splaying and outer retinal thinning in asymptomatic sickle cell patients by spectral-domain optical coherence tomography. Am J Ophthalmol. 2011;151(6):990-994.e991.
- Sambhav K, Grover S, Chalam KV. Temporal thinning in sickle cell retinopathy is associated with diminished perfusion on OCTA and dense scotoma on microperimetry. Retin Cases Brief Rep. 2019;13(4):308-313.
- Stevens TS, Busse B, Lee CB, Woolf MB, Galinos SO, Goldberg MF. Sickling hemoglobinopathies; macular and perimacular vascular abnormalities. Arch Ophthalmol. 1974;92(6):455-463.
- Han IC, Tadarati M, Pacheco KD, Scott AW. Evaluation of macular vascular abnormalities identified by optical coherence tomography angiography in sickle cell disease. Am J Ophthalmol. 2017;177:90-99.
- Ong SS, Linz MO, Li X, Liu TYA, Han IC, Scott AW. Retinal thickness and microvascular changes in children with sickle cell disease evaluated by optical coherence tomography (OCT) and OCT angiography. Am J Ophthalmol. 2020;209:88-98.
- Kotoula M, Papageorgiou E, Xanthou F, Kalampalikis S, Androudi S, Tsironi EE. Choroidal vascular occlusion in a young male patient with sickle cell trait. Int J Ophthalmol. 2018;11(3):528-532.
- Acacio I, Goldberg MF. Peripapillary and macular vessel occlusions in sickle cell anemia. Am J Ophthalmol. 1973;75(5):861-866.
- Knapp JW. Isolated macular infarction in sickle cell (SS) disease. Am J Ophthalmol. 1972;73(6):857-859.
- Liem RI, Calamaras DM, Chhabra MS, Files B, Minniti CP, Thompson AA. Sudden onset blindness in sickle cell disease due to retinal artery occlusion. Pediatr Blood Cancer. 2008;50(3):624-627.
- Chen X, Rahimy E, Sergott RC, et al. Spectrum of retinal vascular diseases associated with paracentral acute middle maculopathy. Am J Ophthalmol. 2015;160(1):26-34.e21.
- Hussnain SA, Coady PA, Stoessel KM. Paracentral acute middle maculopathy: precursor to macular thinning in sickle cell retinopathy. BMJ Case Rep. 2017;2017.
- Ilginis T, Keane PA, Tufail A. Paracentral acute middle maculopathy in sickle cell disease. JAMA Ophthalmol. 2015;133(5):614-616.
- Moriarty BJ, Acheson RW, Condon PI, Serjeant GR. Patterns of visual loss in untreated sickle cell retinopathy. Eye (Lond). 1988;2(Pt 3):330-335.
- Martin GC, Denier C, Zambrowski O, et al. Visual function in asymptomatic patients with homozygous sickle cell disease and temporal macular atrophy. JAMA Ophthalmol. 2017;135(10):1100-1105.
- Farber MD, Jampol LM, Fox P, et al. A randomized clinical trial of scatter photocoagulation of proliferative sickle cell retinopathy. Arch Ophthalmol. 1991;109(3):363-367.
- Rodrigues M, Kashiwabuchi F, Deshpande M, et al. Expression Pattern of HIF-1alpha and VEGF Supports Circumferential Application of Scatter Laser for Proliferative Sickle Retinopathy. Invest Ophthalmol Vis Sci. 2016;57(15):6739-6746.
- Siqueira RC, Costa RA, Scott IU, Cintra LP, Jorge R. Intravitreal bevacizumab (Avastin) injection associated with regression of retinal neovascularization caused by sickle cell retinopathy. Acta Ophthalmol Scand. 2006;84(6):834-835.
- Shaikh S. Intravitreal bevacizumab (Avastin) for the treatment of proliferative sickle retinopathy. Indian J Ophthalmol. 2008;56(3):259.
- Cai CX, Linz MO, Scott AW. Intravitreal Bevacizumab for Proliferative Sickle Retinopathy: A Case Series. J VitreoRet Dis. 2017;2(1):32-38.
- Mitropoulos PG, Chatziralli IP, Parikakis EA, Peponis VG, Amariotakis GA, Moschos MM. Intravitreal ranibizumab for stage IV proliferative sickle cell retinopathy: a first case report. Case Rep Ophthalmol Med. 2014;2014:682583.
- Moshiri A, Ha NK, Ko FS, Scott AW. Bevacizumab presurgical treatment for proliferative sickle-cell retinopathy-related retinal detachment. Retin Cases Brief Rep. 2013;7(3):204-205.
- Zhao Y, Singh RP. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context. 2018;7:212532.
- Chen RW, Flynn HW Jr., Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol. 2014;157(4):870-875.e871.
- Pulido JS, Flynn HW, Jr., Clarkson JG, Blankenship GW. Pars plana vitrectomy in the management of complications of proliferative sickle retinopathy. Arch Ophthalmol. 1988;106(11):1553-1557.
- Williamson TH, Rajput R, Laidlaw DA, Mokete B. Vitreoretinal management of the complications of sickle cell retinopathy by observation or pars plana vitrectomy. Eye (Lond). 2009;23(6):1314-1320.
- Khurmi N, Gorlin A, Misra L. Perioperative considerations for patients with sickle cell disease: a narrative review. Can J Anaesth. 2017;64(8):860-869.
- Benson JM, Therrell BL, Jr. History and current status of newborn screening for hemoglobinopathies. Semin Perinatol. 2010;34(2):134-144.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033-1048.
- Pahl DA, Green NS, Bhatia M, et al. Optical coherence tomography angiography and ultra-widefield fluorescein angiography for early detection of adolescent sickle retinopathy. Am J Ophthalmol. 2017;183:91-98.
- Li J, Bender L, Shaffer J, Cohen D, Ying GS, Binenbaum G. Prevalence and onset of pediatric sickle cell retinopathy. Ophthalmol. 2019;126(7):1000-1006.
- Alam M, Lim JI, Toslak D, Yao X. Differential artery-vein analysis improves the performance of octa staging of sickle cell retinopathy. Transl Vis Sci Technol. 2019;8(2):3.
- Freed J, Talano J, Small T, Ricci A, Cairo MS. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant. 2012;47(12):1489-1498.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. New Engl J Med. 2017;376(9):848-855.
- Green NS, Barral S. Emerging science of hydroxyurea therapy for pediatric sickle cell disease. Pediatr Res. 2014;75(1-2):196-204.
- Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. New Engl J Med. 2019;381(6):509-519.
- Blair HA. Crizanlizumab: first approval. Drugs. 2020;80(1):79-84.