







Tumor masquerade syndromes classically refer to conditions that simulate uveitis but in fact have an underlying neoplastic origin. Conversely, the definition can be expanded to include benign conditions that mimic tumors. Neoplastic processes, inflammatory conditions, and infections can present as masquerade syndromes and can elude even the most experienced retinal specialist. Fortunately, these entities are rare in clinical practice. In one series from a prominent referral center of 853 patients presenting with uveitis, only 2.5% were subsequently diagnosed with a neoplastic masquerade syndrome.1 Delay in diagnosis, in some cases exceeding several years, highlights the difficulty these syndromes pose for clinicians.2 Recognizing masquerade syndromes and differentiating them from their benign counterparts (and vice versa) is of high importance for initiation of prompt and appropriate treatment. The following discussion focuses on 2 notorious masqueraders: lymphoma and amelanotic lesions of the choroid. While not intended to be a comprehensive review of masquerade syndromes, these entities highlight the challenge of determining benign from malignant conditions. Each condition is presented with clinical pearls to aid the retinal physician.
PRIMARY VITREORETINAL LYMPHOMA
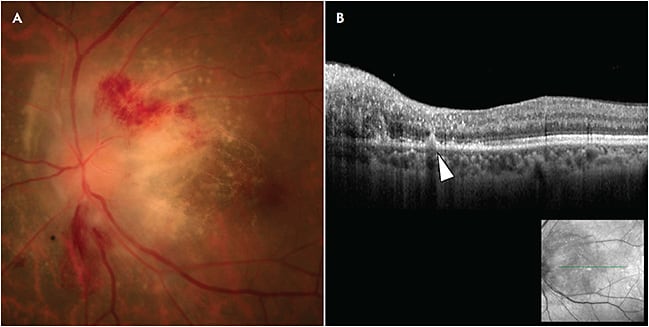
While primary vitreoretinal lymphoma (PVRL) is well known as a masquerade syndrome, this entity continues to pose a diagnostic challenge, and it has an average delay in diagnosis exceeding 1 year from the time of symptom onset.3 Any first episode of posterior uveitis in an older individual should raise suspicion for malignancy. The hallmark features of PVRL are vitreous cells (particularly large, clumped cells and with an “aurora borealis” appearance) and subretinal pigment epithelium (RPE) infiltrates. With classic findings, the diagnosis can be straightforward. Subtle infiltration can be more easily observed on optical coherence tomography (OCT), which can reveal hyper-reflective lymphoma cells in the sub-RPE space and within the retina (Figure 1). Biopsy remains the gold standard for diagnosis; however, the fragility of lymphoma cells and paucity of cells in some samples are problematic. The need for multiple biopsies for initial diagnostic confirmation is common. Fortunately, there have been advances in supportive analytic studies, including flow cytometric immunophenotyping, polymerase chain reaction (PCR) gene rearrangement studies, and analysis of interleukin (IL) levels (an elevated IL10-to-IL6 ratio is supportive of diagnosis). More recently, the MYD88 L265P mutation has been shown to be present in approximately 80% of cases.4 If the MYD88 allele-specific PCR is positive in the context of CD20+ vitreous cells, diagnosis of PVRL can be established. Some centers consider MYD88 mutational analysis to be preferable to standard PCR for monoclonal B-cell rearrangement, because more cells are required for the latter and immunoglobulin heavy-chain gene rearrangement is associated with a high false-negative rate.5 The majority of PVRLs are aggressive, diffuse large B-cell lymphomas (rarely natural-killer cell or T-cell lymphoma). Rapid and accurate diagnosis is critical for initiating prompt treatment and staging to rule out life-threatening central nervous system involvement.
Clinical Pearl for Primary Vitreoretinal Lymphoma
Because many cases of PVRL are initially labeled as “refractory uveitis,” patients are frequently prescribed steroids initially. Whenever possible, steroids should be rapidly tapered and discontinued for several weeks prior to biopsy (due to their lympholytic effect and ability to decrease diagnostic yield). Similarly, staging including magnetic resonance imaging (MRI) of the brain and lumbar puncture (in some cases) should be performed (or repeated) several weeks following discontinuation of steroids.
PRIMARY CHOROIDAL LYMPHOMA
Primary choroidal lymphoma is generally a low-grade malignancy, with extranodal marginal zone lymphoma constituting the majority of cases.6,7 The clinical presentation of multifocal, creamy, white-to-yellow choroidal infiltrates can be a diagnostic challenge. Uveitic conditions such as birdshot chorioretinopathy and sarcoidosis with choroidal involvement, as well as malignancy and particularly choroidal metastases should be considered. The infiltrates in choroidal lymphoma tend to be larger and more randomly distributed than birdshot lesions, which are smaller, more oval-shaped, and have a predilection for the nasal quadrants of the fundus. Indocyanine green (ICG) angiography is particularly useful for characterizing choroidal infiltrates because it helps to delineate the shape and number of lesions, and it can detect subtle, asymmetric involvement in the fellow eye.8 Birdshot chorioretinopathy is more commonly associated with inflammatory features: fine keratic precipitates, cell, flare, and macular edema. Cases of HLA-A29 negative birdshot should raise suspicion for choroidal lymphoma. Sarcoidosis can be differentiated by confirmatory pathology from pulmonary involvement (such as hilar adenopathy) when present, and supportive laboratory testing, including elevated angiotensin-converting enzyme (ACE) and lysozyme levels, can be helpful. Patients with choroidal metastases will frequently have a history of malignancy; however, when the choroid is the presenting site, systemic imaging (MRI, computed tomography [CT], or positron emission tomography [PET]) can help to identify a primary neoplasm.
Clinical Pearl for Primary Choroidal Lymphoma
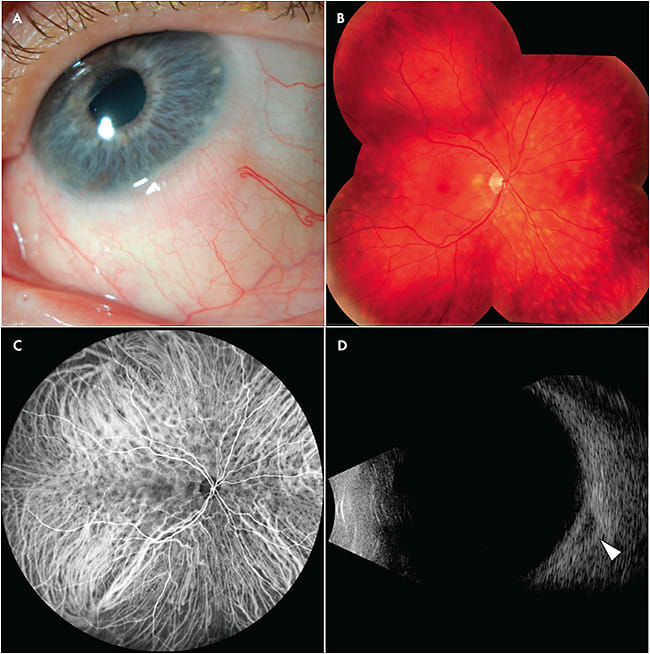
There is an under-recognized and significant overlap between choroidal lymphoma and ocular adnexal lymphoma.9 Careful inspection, including lifting the eyelids to identify epibulbar involvement, is recommended. It has been estimated that more than 70% of uveal lymphoma cases exhibit occult extrascleral extension, therefore B-scan ultrasonography should be performed, both for the apparently involved eye and for the fellow eye. A low-reflective mass located near the optic nerve, is classic and provides a potential biopsy site for diagnostic confirmation (Figure 2).
SCLEROCHOROIDAL CALCIFICATION
Sclerochoroidal calcification is a mimic of more ominous diagnoses, particular choroidal metastases. Patients are often referred in the setting of a known history of malignancy when these multifocal, white-to-yellow, flat to slightly nodular elevations are noted on fundus examination. The lesions are frequently located along the arcades and in the superotemporal quadrant of the fundus.10 Optical coherence tomography reveals suprachoroidal hyporeflective lesions with a table-top configuration. Most cases of sclerochoroidal calcification occur in the elderly and are idiopathic. Occasionally, calcification deposition can be indicative of an underlying imbalance in calcium and phosphate metabolism due to hyperparathyroidism. Rarely, this finding can be associated with renal dysfunction due to Gitelman syndrome or Bartter syndrome. For this reason, laboratory studies including serum calcium, phosphorus, magnesium, parathyroid hormone, and calcitonin are recommended.
Clinical Pearl for Sclerochoroidal Calcification
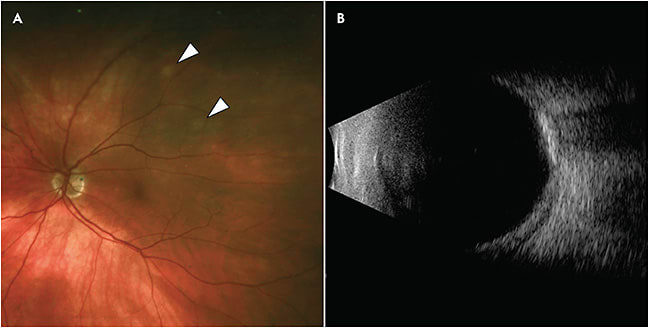
Although these lesions are flat to minimally elevated, ultrasonography is the key to diagnosis. B-scan demonstrates highly reflective lesions with posterior shadowing, confirming the presence of calcium (Figure 3).
NODULAR POSTERIOR SCLERITIS
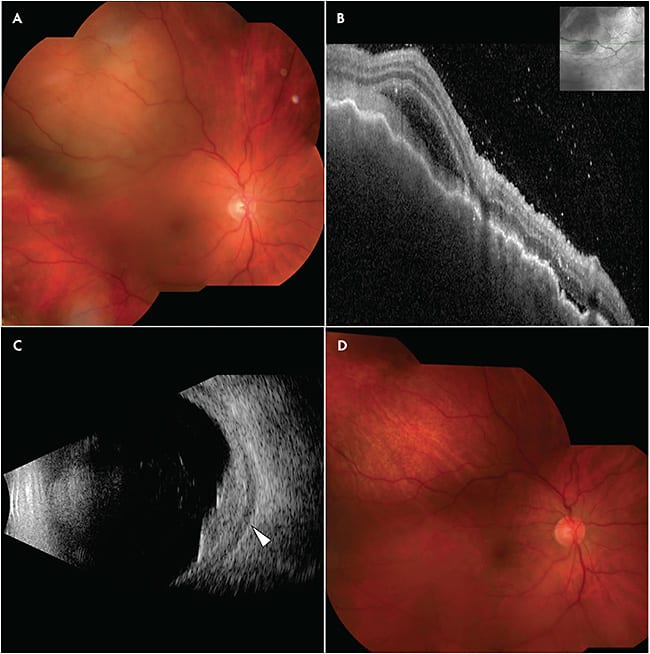
Nodular posterior scleritis poses a particular diagnostic challenge because it presents as an amelanotic choroidal mass, an entity with a broad differential diagnosis including amelanotic melanoma, choroidal metastasis, choroidal lymphoma, circumscribed choroidal hemangioma, choroidal granuloma, and schwannoma.11 Thorough patient history and multimodal imaging are the keys to establishing the correct diagnosis. The medical history should focus on underlying autoimmune disease, vasculitic conditions (particularly rheumatoid arthritis and granulomatosis with polyangiitis), and malignancy. In one large series of 137 patients, posterior scleritis was twice as common in women as in men, and 29% had an associated systemic disease.12 Nodular posterior scleritis most commonly presents as a unilateral, solitary choroidal mass and can be associated with subretinal fluid. Pain with eye movement is common. Although solitary choroidal metastasis can occur, bilateral, multifocal involvement in the setting of a known history of malignancy is more typical. Indocyanine green angiography can readily differentiate choroidal hemangioma, which demonstrates early and avid filling due to its vascular nature. Optical coherence tomography of nodular posterior scleritis will often show an irregular surface contour, in contrast to choroidal melanoma which more commonly has a dome-shaped surface architecture. Hyper-reflective material and vitreous cells overlying the lesion, when present, are more suggestive of an inflammatory etiology (Figure 4). Optical coherence tomography may also demonstrate choroidal folds in 30% of cases of nodular posterior scleritis, a finding not common in choroidal melanoma.13 When there is fluid in Tenon’s space, the classic T-sign on ultrasonography is helpful, however this finding is only present in 36% of cases.13 For choroidal lesions more than 2 mm in thickness, MRI can also facilitate diagnosis. Nodular posterior scleritis typically demonstrates a hypointense signal relative to vitreous for both T1- and T2-weighted imaging.14 In contrast, choroidal melanoma is hyperintense on T1-weighted images and hypointense on T2-weighted imaging.15 Given the strong association with underlying systemic disease, a thorough laboratory workup is warranted, including complete blood count, ACE, lysozyme, QuantiFERON-TB Gold, venereal disease research laboratory test or syphilis antibody, antinuclear antibody, rheumatoid factor, and antineutrophil cytoplasmic antibody.13 Systemic imaging can be helpful to exclude sarcoidosis or a primary neoplasm.
Clinical Pearl for Nodular Posterior Scleritis
Although the majority of cases of nodular posterior scleritis will present with typical findings of scleritis including pain and redness, approximately 25% of cases are painless.13
CHOROIDAL GRANULOMA
As it often appears as an amelanotic mass, choroidal granuloma is a well-known simulator of tumors of the choroid, particularly choroidal metastasis and amelanotic choroidal melanoma.16 Choroidal granuloma is most frequently encountered in association with sarcoidosis and tuberculosis. Rarely, infections such as syphilis, toxocariasis, and Bartonella henselae, among others, can cause granulomas in the choroid. These lesions appear as a yellow-to-white mass in the choroid. There can be associated subretinal fluid. Optical coherence tomography can confirm thickening of the choroid and there may also be overlying disruption of the RPE (Figure 5). Indocyanine green angiography typically shows decreased filling or perfusion corresponding to the clinically observed choroidal mass, excluding the diagnosis of circumscribed choroidal hemangioma, which display early and avid filling. Fluorescein angiography can show optic disc leakage in some cases, which is suggestive of an inflammatory process. A thorough patient history focused on past history of malignancy, age-appropriate cancer screening, prior infection, and travel to areas endemic for tuberculosis are key. Laboratory testing for tuberculosis (QuantiFERON-TB Gold), sarcoidosis (ACE, lysozyme), syphilis (venereal disease research laboratory test or syphilis antibody), toxocariasis (excretory-secretory antigen or Goldmann-Witmer coefficient), and Bartonella henselae (B. henselae IgG and IgM) can be helpful. Systemic imaging can also facilitate diagnosis. In cases where active infection has been excluded, responsiveness to a steroid trial is supportive of the diagnosis of choroidal granuloma and suggests an inflammatory etiology. For particularly challenging cases, fine-needle aspiration biopsy can provide cytologic confirmation of diagnosis.
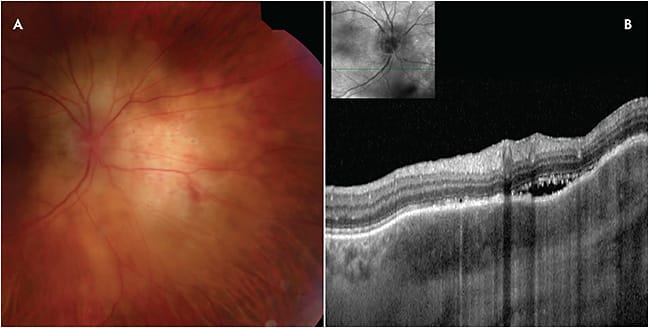
Clinical Pearl for Choroidal Granuloma
While PET can be useful for screening for primary malignancy, it has lower diagnostic specificity for sarcoidosis. Uptake of the fluorodeoxyglucose tracer is readily seen in infectious, inflammatory, and neoplastic conditions. Fluorodeoxyglucose uptake in sarcoidosis is nonspecific, both in intensity and pattern, therefore standard CT chest imaging or combined PET/CT imaging are preferable.17
CONCLUSION
Tumor masquerade syndromes constitute a small percentage of patients seen in clinical practice. Retina specialists should be familiar with these conditions and, in the appropriate setting, should have a high suspicion for neoplastic processes masquerading as uveitis. Similarly, it is important to recognize benign entities that can simulate tumors in order to avoid unnecessary treatments and expensive investigative studies. A detailed medical history, thorough examination, laboratory studies, and use of multimodal ancillary imaging will facilitate diagnosis in the majority of cases. RP
REFERENCES
- Grange LK, Kouchouk A, Dalal MD, et al. Neoplastic masquerade syndromes in patients with uveitis. Am J Ophthalmol. 2013;157(3):526-531.
- Engelhard SB, Aronow ME, Shah CT, Sim AJ, Reddy AK. Malpractice litigation in ocular oncology. Ocul Oncol Pathol. 2018;4(3):135-140.
- Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist. 2011;16(11):1589-1599.
- Raja H, Salomao DR, Viswanatha DS, Pulido JS. Prevalence of Myd88 L265p mutation in histologically proven, diffuse large B-cell vitreoretinal lymphoma. Retina. 2016;36(3):624-628.
- Pulido JS, Johnston PB, Nowakowski GS, Catellino A, Raja H. The diagnosis and treatment of primary vitreoretinal lymphoma: a review. Int J Retina Vitreous. 2018;4:18.
- Aronow ME, Portell CA, Sweetenham JW, Singh AD. Uveal lymphoma: clinical features, diagnostic studies, treatment selection, and outcomes. Ophthalmology. 2014;121(1):334-341.
- Mashayekhi A, Shukla SY, Shields JA, Shields CL. Choroidal lymphoma: clinical features and association with systemic lymphoma. Ophthalmology. 2014;121(1):342-351.
- Reddy AK, Gonzalez MA, Henry CR, Yeh S, Sobbrin L, Albbini TA. Diagnostic sensitivity of indocyanine green angiography for birdshot chorioretinopathy. JAMA Ophthalmol. 2015;133(7):840-843.
- Fuller ML, Sweetenham J, Schoenfield L, Singh AD. Uveal lymphoma: a variant of ocular adnexal lymphoma. Leuk Lymphoma. 2008;49(12):2393-2397.
- Honavar SG, Shields CL, Demirci H, Shields JA. Sclerochoroidal calcification clinical manifestations and systemic associations. Arch Ophthalmol. 2001;119(6):833-840.
- Shibata Y, Kase S, Namba K, Ishida S. A case of nodular posterior scleritis simulating intraocular tumor. Int J Ophthalmol. 2019;12(4):685-688.
- McCluskey PJ, Watson PG, Lightman S, Haybittle J, Restori M, Branley M. Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology. 1999;106(12):2380-2386.
- Agrawal R, Lavric A, Restori M, Pavesio C, Sagoo MS. Nodular posterior scleritis: clinico-sonographic characteristics and proposed diagnostic criteria. Retina. 2016;36(2):392-401.
- DePotter P, Shields CL, Shields JA. Inflammatory disorders. In: DePotter P, Shields JA, Shields CL, eds. MRI of the Eye and Orbit. Philadelphia, PA: JB Lippincott Co; 1995;45-55.
- Houle V, Bélair M, Allaire GS. AIRP best cases in radiologic- pathologic correlation: choroidal melanoma. Radiographics. 2011;31(5):1231-1236.
- Turkoglu EB, Lally SE, Shields CL. Choroidal sarcoid granuloma simulating prostate carcinoma metastasis. Retin Cases Brief Rep. 2017;11 Suppl 1:S226-S228.
- Prabhakar HB, Rabinowitz CB, Gibbons FK, O’Donnell WJ, Shepard JA, Aquino SL. Imaging features of sarcoidosis on MDCT, FDG PET, and PET/CT. AJR Am J Roentgenol. 2008;190(3 Suppl):S1-S6.