







Vogt-Koyanagi-Harada (VKH) disease is characterized as a bilateral non-necrotizing granulomatous pan-uveitis that presents in various stages with extraocular findings. The spectrum of clinical presentation includes both intraocular manifestations such as exudative retinal detachment (RD) as well as extraocular findings such as vitiligo, poliosis, auditory changes, and neurologic deficits.1 Depending on the point in the disease course in which patients present for evaluation, diagnosis may be difficult, because clinical findings may vary greatly. Furthermore, a standardized diagnostic criterion is lacking, as multiple algorithms are currently utilized with varying success.2 In this article, we explore the recent studies and reports that seek to elucidate the pathophysiology behind this disease as well as developments to improve diagnosis and treatment.
PATHOPHYSIOLOGY
Although the specific disease mechanism of VKH is still not well understood, mounting evidence suggests that the disease is mediated by an activation of T lymphocytes against antigens within melanocytes. More specifically, activated T helper (Th) cells react toward specific melanocytic proteins such as tyrosinase to initiate downstream proinflammatory signaling cascades to trigger an autoimmune response.3-5 Further studies are necessary to elucidate the secondary pathways of Th cell activation, which may provide invaluable pharmacologic targets for future therapies.
A genetic predisposition to VHK is apparent, because the prevalence of the disease varies significantly by ethnicity. While groups including Asians, Hispanics, and Native Americans are more frequently affected, this disease is less commonly found in those of white and sub-Saharan African descent.6 Studies evaluating the genetics of the human leukocyte antigen (HLA) system, a class II major histocompatibility complex, found a high association between VKH and the HLA-DRB1*04 allele, with suballeles 0404, 0405, and 0410 conferring a higher risk.4,7-9 It is tempting to assume that genetic predisposition explains the variance in patient susceptibility to developing VKH, but this association was found to be different between various ethnic groups, suggesting other contributing factors may be involved.9
One intriguing possibility is the existence of an environmental trigger. It has been postulated that VKH is triggered in genetically predisposed patients by an infectious agent, most likely a virus. Numerous viruses have been implicated in triggering VKH, including Epstein-Barr10 and cytomegalovirus.11 There has also been a case report implicating hepatitis B vaccination as a trigger for VKH.12 The proposed mechanism is that a viral infection may stimulate the production of Th cells that cross-react with melanocytic antigens such as tyrosinase through the mechanism of molecular mimicry.11 Further studies are necessary to elucidate the molecular triggers and downstream pathways involved in the autoimmune response.
CLINICAL PRESENTATION
VKH classically presents as 4 distinct phases: prodromal, acute uveitic, convalescent, and chronic recurrent.3 The prodromal phase initially presents as a nonspecific viral-like illness; as such, affected patients do not often present to ophthalmology for evaluation. Its duration lasts for only a few days and is marked by headaches, fever, orbital pain, meningismus, and hearing loss.8 After the onset of initial symptoms, patients may also experience photophobia. Lumbar puncture to obtain cerebrospinal fluid during this phase reveals pleocytosis.
Once the patient progress to the acute uveitic phase, ocular manifestations become prevalent, including diffuse choroiditis with exudative RDs, subretinal precipitates, optic disc swelling, and anterior and/or vitreous inflammatory cells (Figure 1). Often, patients present to ophthalmology during this phase due to significant loss of vision.

In the convalescence phase, integumentary findings manifest, such as poliosis, vitiligo, and alopecia.8 Ophthalmic findings include depigmentation of the choroid that manifests as a “sunset glow fundus” appearance, chorioretinal scarring with scattered clumping of the retinal pigment epithelium (RPE), and depigmentation of the limbus, which is commonly referred to as “Sigiura’s sign.”8-10 Finally, the chronic recurrent phase refers to recurrent episodes of anterior and/or posterior uveitis that may last for years.
DIAGNOSIS
Although the diagnosis of VKH is based on clinical findings alone, there does not exist a standard diagnostic criterion. Numerous criteria have been proposed, including the American Uveitis Society criteria,13 Sugiura criteria,14 and the Revised Diagnostic Criteria (RDC; Table 1) set forth by the First International Workshop on VKH Disease.6 Of these, the RDC is presently the most widely used and is highly sensitive and specific compared with the other 2 criteria.15-18 In a study applying the RDC to known VKH patients, only 12% were classified with the complete form of the disease, whereas 71% and 9% were classified to have incomplete and probable VKH, respectively.16
| A. Complete VKH Disease | ||||
| 1. Bilateral ocular involvement (a or b must be met, depending on the stage of disease when the patient is examined) | ||||
| a. Early manifestations of disease | ||||
| 1) There must be evidence of diffuse choroiditis (with or without anterior uveitis, vitreous inflammatory reaction, or optic disc hyperemia), which may manifest as one of the following: | ||||
| a) Focal areas of subretinal fluid, or | ||||
| b) Bullous serous retinal detachments | ||||
| 2) With equivocal fundus findings, both of the following must be present as well: | ||||
| a) Focal areas of delay in choroidal perfusion, multifocal areas of pin-point leakage, large placoid areas of hyperfluorescence, pooling within subretinal fluid, and optic nerve staining (listed in order of sequential appearance) by fluorescein angiography, and | ||||
| b) Diffuse choroidal thickening, without evidence of posterior scleritis by ultrasonography | ||||
| b. Late manifestations of disease | ||||
| 1) History suggestive of prior presence of findings from 1a, and either both (2) and (3) below, or multiple signs from (3): | ||||
| 2) Ocular depigmentation (either of the following manifestations is sufficient): | ||||
| a) Sunset glow fundus, or | ||||
| b) Sugiura sign | ||||
| 3) Other ocular signs: | ||||
| a) Nummular chorioretinal depigmented scars, or | ||||
| b) Retinal pigment epithelium clumping and/or migration, or | ||||
| c) Recurrent or chronic anterior uveitis | ||||
| 2. Neurologic/auditory findings (may have resolved by time of examination) | ||||
| a. Meningismus (malaise, fever, headache, nausea, abdominal pain, stiffness of the neck and back, or a combination of these factors; headache alone is not sufficient to meet the definition of meningismus, however), or | ||||
| b. Tinnitus, or | ||||
| c. Cerebrospinal fluid pleocytosis | ||||
| 3. Integumentary finding (not preceding onset of central nervous system or ocular disease) | ||||
| a. Alopecia, or | ||||
| b. Poliosis, or | ||||
| c. Vitiligo | ||||
| B. Incomplete VKH Disease (Point 1 and either 2 or 3 must be present) | ||||
| 1. Bilateral ocular involvement as defined for complete VKH disease | ||||
| 2. Neurologic/auditory findings as defined for complete VKH disease above, or | ||||
| 3. Integumentary findings as defined for complete VKH disease above | ||||
| Probable VKH Disease | ||||
| 1. Bilateral ocular involvement as defined for complete VKH disease above | ||||
| NOTE: IN ALL CASES, THERE SHOULD NOT BE A HISTORY OF PENETRATING OCULAR INJURY OR SURGERY PRECEDING THE INITIAL ONSET OF UVEITIS AND THERE SHOULD BE NO CLINICAL OR LABORATORY EVIDENCE SUGGESTIVE OF OTHER OCULAR DISEASE CRITERIA (MODIFIED FROM READ ET AL, 20016). | ||||
Several limitations of the RDC may explain the low proportion of known VKH patients diagnosed with complete VKH. First, although studies have shown that VKH disease may manifest in some patients as an isolated intraocular inflammation,19 the RDC does not allow for a definitive diagnosis without any extraocular manifestations. Although traditionally a multisystemic disease, VKH has been reported to manifest with isolated ocular involvement (previously termed Harada disease)20 in up to 45% of patients in the acute phase and in up to 58% of those presenting with chronic disease.17 Alternatively, patients can also present with an incomplete form of the disease with bilateral ocular involvement and either integumentary or neurologic manifestations, including auditory findings such as tinnitus and hearing impairment.6
Given the reliance of the RDC on extraocular findings for a diagnosis of “complete VKHD,” one must give consideration to the value of ancillary testing and systemic physical exams in search of other cardinal features. Seguira et al previously described diagnostic criteria with mandated cerebrospinal fluid analysis,21 in line with a more invasive approach to discovering extraocular manifestation; however, the system is rarely used outside of Japan, potentially because of this additional mandate which is useful namely in the acute stage of the disease.22 In general, there appears to be substantial utility in pursuing additional diagnostics such as lumbar puncture and audiologic testing to identify extraocular findings, although this testing should be performed on the basis of clinical need.23
Another limitation to the RDC is that it depends heavily upon the identification of fundus features, which can be difficult to observe in the late phase of VKH disease due to the presence of severe media opacities. Finally, fundus features may also be atypical in patients who have previously been treated with corticosteroids, thus complicating the initial diagnosis using the RDC.24 Other groups have put forth additional diagnostic criteria, including advancing diagnostics (ie, changes on full-field electroretinography and enhanced-depth optical coherence tomography [ED-OCT]), but these require further validation.2,15,25
IMAGING
Aside from direct visualization through clinical exam, retinal imaging is essential to diagnosis and monitoring of VKH. Optical coherence tomography provides high-resolution imaging of the subretinal fluid associated with serous RDs in eyes with acute VKH alone, and can indicate resolution during the course of successful treatment (Figure 2).26,27 Enhanced-depth OCT uses a longer wavelength to enhance visualization of the choroid. Previous studies have shown that choroidal thickness derived from this method is increased in active VKH disease, decreased with effective treatment, and was thinner in eyes with chronic disease.28,29
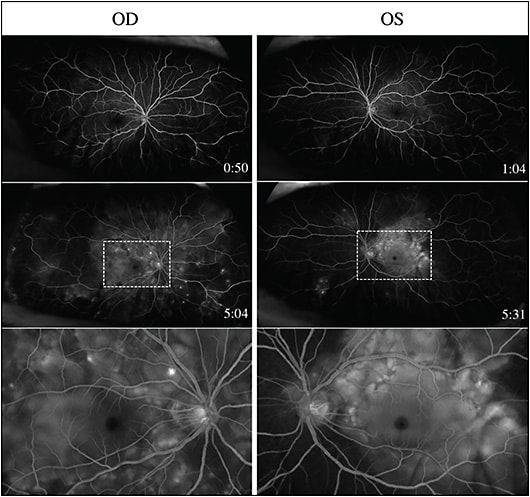
Fluorescein angiography (FA) is widely used to image active VKH and often demonstrates multifocal pinpoint areas of leakage throughout the fundus along with subretinal pooling of fluorescein in areas of exudative RD (Figure 3).30,31 To further monitor changes in choroidal circulation during the course of VKH, indocyanine green angiography (ICGA) can be utilized to show hypofluorescent spots in both active disease and chronic stromal scarring within the choroid.32 Furthermore, OCT angiography is a noninvasive modality used to view retinal and choroidal vascular flow without dye injection, and it allows for visualization and detailed evaluation of retinal and choroidal vascularity to monitor VKH patients over time.33-36

Multispectral image analysis is a novel technique using individual monochromatic light emitting diodes (LEDs) in an expanded range of wavelengths between 550 nm and 850 nm to create discrete spectral slices of the retina based on absorption of specific wavelengths.37 It can image melanin, which absorbs longer wavelengths (red and infrared) to reveal deep retinal and subretinal architecture. This imaging modality has been utilized as a noninvasive method to better evaluate the RPE of VKH patients for diagnosis compared to traditional methods such as OCT, FA, and fundus photography.38 Further studies are needed to optimize and integrate this methodology into the standardized workup and evaluation of patients.
Finally, VKH patients can be monitored through non-traditional imaging modalities for intraocular disease. Studies have found that high-resolution magnetic resonance imaging (MRI) of the eye and orbit can elucidate the extent of the disease. Specifically, MRI was able to sensitively detect thickening of the choroid as well as tenons capsule in VKH patients on T1- and T2-weighted images.39 Furthermore, MRI detected perineural infiltrative changes along the optic nerve with meningeal enhancement of adjacent cerebral structures.40,41 The utility of this imaging modality for ophthalmic practice remains limited; however, it provides an intriguing method to monitor disease burden.
TREATMENT
The mainstay of therapy for VKH disease remains immediate corticosteroid therapy in the acute phase.42 Studies have found no difference in clinical outcomes between oral and intravenous corticosteroids; however, recurrent inflammation and declining visual outcomes were more often associated with withdrawing oral therapy within 6 months of initiation.43 Periocular and intravitreal corticosteroid therapies have also been shown to be successful in controlling ocular inflammation in VKH.44,45
Supplementation with immunomodulatory therapy (IMT) may be necessary for patients with ocular inflammation associated with VKH that is not adequately maintained with corticosteroid therapy, or if the amount of necessary corticosteroid therapy is unacceptably high.46-48 Other studies have found that using IMT as a first-line treatment in combination with systemic corticosteroid therapy may delay or prevent the conversion to the more chronic and recurrent phases of VKH.49 While various classes of IMTs have been successfully used to treat VKH patients, care must be taken to monitor for unwanted side effects of these medications, including aplastic anemia and liver dysfunction.50 Comanaging VKH patients in need of IMT with a uveitis-trained ophthalmologist or rheumatologist is advised.
Regardless of the pharmacologic agent selected to treat newly diagnosed VKH patients, multiple studies have found that earlier therapeutic initiation has been linked to improved long-term visual outcomes.51,52 An interesting notion of a therapeutic “window of opportunity” exists in the initial onset of acute VKH disease where aggressive treatment can lead to substantial clinical improvements and may prevent its progression to chronic and recurrent forms.53 Further studies are needed to understand the pathophysiology of disease so that more targeted therapeutics can be developed.
CONCLUSION
Although the clinical features of VKH have been well documented, the understanding of its pathogenesis remains unclear. Numerous basic science and clinical studies are under way to better understand the disease mechanisms so that more effective therapies and treatment strategies can be devised for patients suffering from this devastating condition. Because early initiation of therapy is crucial to controlling the course of VKH in patients, clinicians must be diligent in their diagnosis and treatment strategies. RP
REFERENCES
- Burkholder BM. Vogt-Koyanagi-Harada disease. Curr Opin Ophthalmol. 2015;26(6):506-511.
- Yang P, Zhong Y, Du L, et al. Development and evaluation of diagnostic criteria for Vogt-Koyanagi-Harada disease. JAMA Ophthalmol. 2018; in print.
- Imai Y, Sugita M, Nakamura S, Toriyama S, Ohno S. Cytokine production and helper T cell subsets in Vogt-Koyanagi-Harada’s disease. Curr Eye Res. 2001;22(4):312-318.
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase family proteins are antigens specific to Vogt-Koyanagi-Harada disease. J Immunol. 2000;165(12):7323-7329.
- Gocho K, Kondo I, Yamaki K. Identification of autoreactive T cells in Vogt-Koyanagi-Harada disease. Invest Ophthalmol Vis Sci. 2001;42(9):2004-2009.
- Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131(5):647-652.
- Shindo Y, Inoko H, Yamamoto T, Ohno S. HLA-DRB1 typing of Vogt-Koyanagi-Harda’s disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Ophthalmol. 1994;78(3):223-226.
- Islam SM, Numaga J, Fujino Y, et al. HLA class II genes in Vogt-Koyanagi-Harada disease. Invest Ophthalmol Vis Sci. 1994;35(11):3890-3896.
- Shi T, Lv W, Zhang L, Chen J, Chen H. Association of HLA-DR4/HLA-DRB*04 with Vogt-Koyanagi-Harada disease: a systematic review and meta-analysis. Sci Rep. 2014;4:6887.
- Bassili SS, Peyman GA, Gebhardt BM, Daun M, Ganiban GJ, Rifai A. Detection of Epstein-Barr virus DNA by polymerase chain reaction in the vitreous from a patient with Vogt-Koyanagi-Harada Syndrome. Retina. 1996;16(2):160-161.
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Cross-reaction between tyrosinase peptides and cytomegalovirus antigen by T cells from patients with Vogt-Koyanagi-Harada disease. Int Ophthalmol. 2007;27(2-3):87-95.
- Sood AB, O’Keefe G, Bui D, Jain N. Vogt-Koyanagi-Harada disease associated with hepatitis B vaccination. Ocul Immunol Inflamm. 2018; in print.
- Snyder DA, Tessler HH. Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1980;90(1):69-75.
- Sugiura S. Some observations on uveitis in Japan, with special reference to Vogt-Koyanagi-Harada and Behçet disease. Nippon Ganka Gakkai Zasshi. 1976;80(11):1285-1326.
- da-Silva FT, Damico FM, Marin ML, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: considerations on the different disease categories. Am J Ophthalmol. 2009;147(2):339-345.
- Kitamura M, Takami K, Kitaichi N, et al. Comparative study of two sets of criteria for the diagnosis of Vogt-Koyanagi-Harada’s disease. Am J Ophthalmol. 2005;139(6):1080-1085.
- Rao NA, Sukavatcharin S, Tsai JH. Vogt-Koyanagi-Harada disease diagnostic criteria. Int Ophthalmol. 2007;27(2-3):195-199.
- Read RW, Rao NA. Utility of existing Vogt-Koyanagi-Harada syndrome diagnostic criteria at initial evaluation of the individual patient: a retrospective analysis. Ocul Immunol Inflamm. 2000;8(4):227-234.
- Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt-Koyanagi-Harada disease. Ophthalmology. 2010;117(3):591-599.
- Cowper AR. Harada’s disease and Vogt-Koyanagi syndrome; uveoencephalitis. AMA Arch Ophthalmol. 1951;45(4):367-376.
- Suguira S. Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1978;22:9-35.
- Li F, Yang P, Liu X, Wang C, Hou S, Kijlstra A. Upregulation of interleukin 21 and promotion of interleukin 17 production in chronic or recurrent Vogt-Koyanagi-Harada disease. Arch Ophthalmol. 2010;128(11):1449-1454.
- Al-Dousary S. Auditory and vestibular manifestations of Vogt-Koyanagi-Harada disease. J Laryngol Otol. 2011;125(2):138-141.
- Chee SP, Jap A, Bacsal K. Prognostic factors of Vogt-Koyanagi-Harada disease in Singapore. Am J Ophthalmol. 2009;147(1):154-161.
- Yang P, Du L, Ye Z. How to deal with uveitis patients? Curr Mol Med. 2018;17(7):468-470.
- Ishihara K, Hangai M, Kita M, Yoshimura N. Acute Vogt-Koyanagi-Harada disease in enhanced spectral-domain optical coherence tomography. Ophthalmology. 2009;116(9):1799-1807.
- Tsujikawa A, Yamashiro K, Yamamoto K, Nonaka A, Fujihara M, Kurimoto Y. Retinal cystoid spaces in acute Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 2005;139(4):670-677.
- Nakai K, Gomi F, Ikuno Y, et al. Choroidal observations in Vogt-Koyanagi-Harada disease using high-penetration optical coherence tomography. Graefes Arch Clin Exp Ophthalmol. 2012;250(7):1089-1095.
- Takahashi H, Takase H, Ishizuka A, et al. Choroidal thickness in convalescent Vogt-Koyanagi-Harada disease. Retina. 2014;34(4):775-780.
- Attia S, Khochtali S, Kahloun R, et al. Clinical and multimodal imaging characteristics of acute Vogt-Koyanagi-Harada disease unassociated with clinically evident exudative retinal detachment. Int Ophthalmol. 2016;36(1):37-44.
- Chee SP, Jap A, Cheung CM. The prognostic value of angiography in Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2010;150(6):888-893.
- Knecht PB, Mantovani A, Herbort CP. Indocyanine green angiography-guided management of Vogt-Koyanagi-Harada disease: differentiation between choroidal scars and active lesions. Int Ophthalmol. 2013;33(5):571-577.
- Giannakouras P, Andreanos K, Giavi B, Diagourtas A. Optical coherence tomography angiography: employing a novel technique for investigation in Vogt-Koyanagi-Harada disease. Case Rep Ophthalmol. 2017;8(2):362-369.
- Aggarwal K, Agarwal A, Deokar A, et al. Distinguishing features of acute Vogt-Koyanagi-Harada disease and acute central serous chorioretinopathy on optical coherence tomography angiography and en face optical coherence tomography imaging. J Ophthalmic Inflamm Infect. 2017;7(1):3.
- Aggarwal K, Agarwal A, Mahajan S, et al. The role of optical coherence tomography angiography in the diagnosis and management of acute Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm. 2018;26(1):142-153.
- Agrawal R, Li LK, Nakhate V, Khandelwal N, Mahendradas P. Choroidal vascularity index in Vogt-Koyanagi-Harada disease: an EDI-OCT derived tool for monitoring disease progression. Transl Vis Sci Technol. 2016;5(4):7.
- Calcagni A, Gibson JM, Styles IB, Claridge E, Orihuela-Espina F. Multispectral retinal image analysis: a novel non-invasive tool for retinal imaging. Eye (Lond). 2011;25(12):1562-1569.
- Huang G, Peng J, Ye Z, Kijlstra A, Zhang D, Yang P. Multispectral image analysis in Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2017;In Print.
- Ando T, Kato H, Mochizuki K, Ozawa K, Goshima S, Matsuo M. MR findings of the orbit in patients with Vogt-Koyanagi-Harada disease. Neuroradiology. 2018;60(4):421-426.
- Han HJ, Kim HY, Park JH, Lee EJ, Kim DG, Shin DI. Magnetic resonance imaging of pachymeningeal enhancement in Vogt-Koyanagi-Harada disease. Neurol Sci. 2010;31(6):785-788.
- Lohman BD, Gustafson CA, McKinney AM, Sarikaya B, Silbert SC. MR imaging of Vogt-Koyanagi-Harada syndrome with leptomeningeal enhancement. AJNR Am J Neuroradiol. 2011;32(9):E169-E171.
- Read RW, Yu F, Accorinti M, et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2006;142(1):119-124.
- Lai TY, Chan RP, Chan CK, Lam DS. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease. Eye (Lond). 2009;23(3):543-548.
- Hosoda Y, Hayashi H, Kuriyama S. Posterior subtenon triamcinolone acetonide injection as a primary treatment in eyes with acute Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2015;99(9):1211-1214.
- Latronico ME, Rigante D, Caso F, et al. Bilateral dexamethasone intravitreal implant in a young patient with Vogt-Koyanagi-Harada disease and refractory uveitis. Clin Rheumatol. 2015;34(6):1145-1148.
- O’Keefe GA, Rao NA. Vogt-Koyanagi-Harada disease. Surv Ophthalmol. 017;62(1):1-25.
- Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130(4):492-513.
- Couto C, Schlaen A, Frick M, et al. Adalimumab treatment in patients with Vogt-Koyanagi-Harada disease. Ocul Immunol Inflamm. 2018;26(3):485-489.
- Abu-El-Asrar AM, Dosari M, Hemachandran S, Gikandi PW, Al-Muammar A. Mycophenolate mofetil combined with systemic corticosteroids prevents progression to chronic recurrent inflammation and development of ‘sunset glow fundus’ in initial-onset acute uveitis associated with Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2017;95(1):85-90.
- Urzua CA, Velasquez V, Sabat P, et al. Earlier immunomodulatory treatment is associated with better visual outcomes in a subset of patients with Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2015;93(6):e475-e480.
- Sakata VM, da-Silva FT, Hirata CE, et al. High rate of clinical recurrence in patients with Vogt-Koyanagi-Harada disease treated with early high-dose corticosteroids. Graefes Arch Clin Exp Ophthalmol. 2015;253(5):785-790.
- Chee SP, Jap A, Bacsal K. Spectrum of Vogt-Koyanagi-Harada disease in Singapore. Int Ophthalmol. 2007;27(2-3):137-142.
- Herbort-Jr CP, Abu-El-Asrar AM, Takeuchi M, et al. Catching the therapeutic window of opportunity in early initial-onset Vogt-Koyanagi-Harada uveitis can cure the disease. Int Ophthalmol. 2018; in print.