







The US Food and Drug Administration (FDA) approval of voretigene neparvovec (Luxturna; Spark Therapeutics) heralds the advent of a new era in the management of inherited retinal degenerations (IRDs). Retinal physicians serve on the front lines in identifying patients with IRDs and thus should stay abreast of these exciting developments in the field.
Although we don’t typically have resources for comprehensive evaluation of IRD patients in our injection-oriented clinics, we do have a number of useful tools at our disposal. This article offers a refresher on key clinical pearls in approaching patients with IRDs, for retinal physicians who do not routinely care for this population.
Nieraj Jain, MD, is an assistant professor of ophthalmology in the department of Vitreoretinal Surgery and Ophthalmic Genetics at Emory University in Atlanta, Georgia. Dr. Jain reports no financial conflicts of interest related to this article.
ASSESSING DISEASE PHENOTYPE IN YOUR GENERAL RETINA CLINIC
Know the “Common” IRDs
In a field of zebras, it helps to know the relative prevalence of various IRDs. Stargardt dystrophy and other ABCA4-mediated diseases, for instance, have an estimated prevalence of approximately 1 in 10,000 individuals in the United States. ABCA4-mediated degeneration is likely the most common monogenic IRD in the United States. In Stone’s recent paper of 1,000 consecutive families with hereditary retinal disease, ABCA4-related disease accounted for 173 cases.1 This was more than twice as common as the second most common genetic disease (USH2A-mediated degeneration) and outnumbered RPE65-mediated disease nearly 60 to 1. Retinitis pigmentosa (RP), which is associated with mutations in many different genes, has an estimated prevalence of 1 in 4,000 US individuals.
Start With the History
Patients with IRDs often present with late-stage manifestations of a disease process that began in childhood. Diseases that initially impact one cell type, such as the rod photoreceptors in RP, ultimately lead to degeneration in neighboring photoreceptors and the retinal pigment epithelium (RPE), making it difficult to categorize the disease based on examination alone.
Questions regarding the initial symptoms, duration of symptoms, and rate of progression can help localize sites of initial disease. We sometimes forget to ask the easy questions, such as, “Do you have difficulty seeing in the dark?” Other classic questions, such as, “Can you see the stars?” or “Can you find your seat in a movie theatre?” and “Do you see better outdoors in sunlight?” address symptoms of nyctalopia, prolonged dark adaptation, and hemeralopia that help differentiate between rod and cone–mediated disease.
The subjective rate of symptomatic progression is particularly important in differentiating hereditary vs more rapidly progressing acquired retinopathies such as autoimmune retinopathy. These historical elements continue to play a valuable role in the diagnostic work-up even in the present era of advanced imaging and molecular diagnostics.
Assess Refractive Error
We routinely assess refractive error in the Emory Ophthalmic Genetics clinic as it can help build the case for certain types of hereditary disease. Hyperopia is present in such diseases as Leber congenital amaurosis (LCA) (note that RPE65-associated LCA may be associated with myopia) and achromatopsia. High myopia is often present in RP, congenital stationary night blindness, and hereditary vitreoretinopathies. A quick trip to the autorefractor can bolster your suspicion of an IRD in these patients.
Identify Key Imaging Findings
Spectral-domain optical coherence tomography (SDOCT) and widefield fundus autofluorescence (FAF) imaging, both of which are fairly ubiquitous technologies in retina clinics, play a central role in the evaluation of IRD patients.
Characteristic features on SDOCT imaging include selective loss of photoreceptor layers and/or RPE that is often symmetric about the fovea and symmetric between the eyes. The hallmark OCT finding in RP is the gradual, tapering loss of the perifoveal outer layers (Figure 1A). There are few acquired diseases that cause such selective outer retinal changes in the absence of prominent collateral damage to the inner retina and RPE.

In contrast to RP, primary choroidal or RPE-based diseases typically feature abrupt transition zones between healthy and diseased retina, often accompanied by outer retinal tubulations (Figure 1B).
Widefield FAF imaging is an essential tool in revealing the nature and extent of degeneration and is particularly helpful in the absence of electroretinography (ERG) testing. This modality can be acquired in almost any patient, including young toddlers with nystagmus, and often reveals disease that is more extensive than appreciated on funduscopic exam.
Hallmark FAF features in hereditary retinopathies are the presence of disease that progresses about a central fulcrum at the fovea (although not necessarily symmetric between superior/inferior and nasal/temporal periphery) and symmetry in findings between eyes (Figure 2A). This pattern of FAF changes is in contrast to some acquired pigmentary retinopathies, such as prior chorioretinitis, chronic/multifocal central serous retinopathy, trauma, and TORCH (Toxoplasmosis, Other [syphilis, varicella zoster, parvovirus B19], Rubella, Cytomegalovirus, Herpes) infections, such as congenital rubella (Figure 2B).
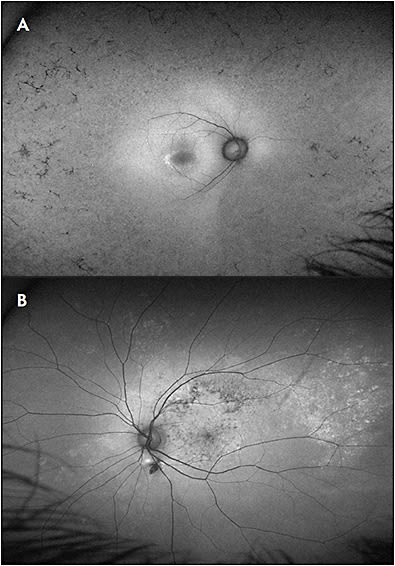
The FAF pattern in RP is typically notable for midperipheral hypoautofluorescence that initially spares the far periphery, mirroring the extent of the ring scotoma that is found on visual field testing. There is typically a hyperautofluorescent macular ring that corresponds to a loss of outer retinal bands that is evident on OCT imaging (Figure 2A). This finding of a hyperautofluorescent ring or line is seen in many conditions at margins of active disease progression between healthy and diseased photoreceptors. Lastly, FAF imaging in RP highlights the attenuation of vessels that is seen in widespread retinal degenerations.
In Stargardt dystrophy, FAF imaging is particularly helpful in identifying highly autofluorescent lipofuscin-containing flecks, with a ring of peripapillary sparing of normal autofluorescence (AF) (Figure 3). Although this finding of peripapillary sparing is not pathognomic for ABCA4-mediated degeneration, it is highly suggestive of a hereditary etiology of disease.

Of note, given some concerns regarding phototoxicity in compromised eyes with short-wavelength AF, we advocate employing longer-wavelength AF devices or reduced-illumination short-wavelength FAF, particularly if patients will undergo serial imaging.2
The clinical utility of OCT angiography imaging in this population is a subject of ongoing inquiry.3 It is particularly helpful in identifying choroidal neovascularization in diseases such as vitelliform dystrophies, where subretinal fluid is often present in the absence of a choroidal neovascular (CNV) membrane.
Treat What You Can
The standard refrain from many IRD specialists is for patients to avoid smoking, wear sunglasses when outdoors, eat well, and exercise. There is currently no consensus regarding optimal diet or nutritional supplementation protocol in patients with IRDs.
In a classic study, high-dose vitamin A supplementation (15,000 IU/day) slowed the rate of decline in flicker ERG amplitudes in patients with RP but had no significant impact on visual acuity or visual field progression.4 Some authors have questioned the study design and interpretation and have pointed to potential toxicities from high-dose vitamin A supplementation.5 Other studies have evaluated supplementation with the omega-3 fatty acid DHA and antioxidant vitamins in RP, although there remains no consensus regarding the clinical impact of supplements. Of note, patients with ABCA4-related disease are advised to avoid excessive vitamin A intake, as animal studies suggest that this may accelerate lipofuscin deposition.
Cystoid macular edema (CME) often manifests in eyes with IRDs and is treatable by a variety of approaches. Topical and oral carbonic anhydrase inhibitors (CAIs) yield anatomic and functional benefits in some patients, although the long-term clinical significance of these gains is not clear. Nonetheless, CAIs are widely prescribed for IRD-associated CME. We typically favor topical therapy for its relatively benign side-effect profile. In refractory cases, we transition to oral acetazolamide. A number of other therapies, including corticosteroids and anti-VEGF agents, have yielded mixed results. All approaches are frustrated by the presence of nonresponders and recurrences.
Retina specialists should be attuned to the risk of CNV membrane formation in certain IRDs. Patients with Best disease are at particularly high risk. One study reported a period prevalence of 5.7%, generally in younger patients, although this may be an underestimate.6 Of note, CNV can often be difficult to discern in the presence of the vitelliform material and subretinal fluid that is typically present in Best disease. OCT angiography is particularly helpful in these cases. When present, CNV appears to have a relatively milder natural history in Best disease as compared with AMD and is typically highly responsive to anti-VEGF therapy when treated early.
CNV can also form in other IRDs, particularly macular dystrophies such as Stargardt disease and Sorsby’s dystrophy, as well as in chorioretinal degenerations such as choroideremia and Bietti dystrophy. Fortunately, data suggest that a light-touch approach such as an as-needed treatment regimen may be well suited to the management of CNV in Best dystrophy and many other hereditary retinopathies.
Lastly, many patients with IRDs develop early-onset cataract. These eyes present unique challenges to the cataract surgeon, and retinal physicians should be generally more conservative in initiating referral for cataract extraction. Some of the risks of surgery in these patients include intraoperative phototoxicity, zonular instability, CME, and postoperative capsular phimosis.
Know the “Can’t Miss” Treatable Diagnoses
RPE65-mediated degeneration is becoming a “can’t miss” diagnosis. The spectrum of RPE65-associated phenotypes ranges from LCA to RP, making it difficult to diagnose on clinical grounds alone. Patients with LCA typically present in infancy with nystagmus, night blindness, and severely attenuated or nonrecordable ERG responses. RPE65-mediated degeneration can be differentiated from some of the other genetic causes of LCA by lack of photophobia and myopia in some cases.7 At early stages, retinal structure is fairly intact on OCT imaging.
Gyrate atrophy is a rare metabolic disorder resulting from deficiency in ornithine aminotransferase, characterized by childhood myopia, nyctalopia, and progressive chorioretinal degeneration. This disorder, which is not necessarily detected on newborn metabolic screens, can be identified by characteristic funduscopic findings of scalloped areas of peripheral chorioretinal atrophy. Patients may benefit from dietary modification with arginine restriction and vitamin B6 supplementation.
Kearns-Sayre is a rare mitochondrial disorder of childhood characterized by bilateral ptosis, chronic progressive external ophthalmoplegia, pigmentary retinopathy, and heart block. Early recognition can lead to potentially life-saving cardiac interventions.
Autoimmune retinopathy, including both paraneoplastic and nonparaneoplastic subtypes, can be difficult to differentiate from hereditary retinopathies. Key distinguishing features include a more rapid rate of progression, generally later age of onset, relative lack of bone spicule pigment changes, and relative asymmetry. Antiretinal antibody testing can aid in diagnosis. Unfortunately, there are many exceptions to these findings, and this condition remains difficult to diagnose even with all of the tools available in a traditional ophthalmic genetics clinic.
Syphilis is another treatable cause of acquired retinal degeneration that has numerous clinical phenotypes. Both congenital and acquired syphilitic retinopathy may be mistaken for an IRD. Syphilitic outer retinopathy may be mistaken for macular dystrophies such as occult macular dystrophy. We often assess treponemal serologies in atypical retinopathies.
A number of drug-associated retinal degenerations can resemble hereditary disease. Some of the more common drug-associated degenerations include those due to hydroxychloroquine (including both the classic paracentral pattern as well as the pericentral pattern in Asian patients), antimalarials, the antipsychotic thioridazine, and a variety of drug-induced crystalline maculopathies. Lastly, the interstitial cystitis drug pentosan polysulfate sodium is associated with a pigmentary maculopathy resembling pattern dystrophy.8
Be Cognizant of Our Limitations
Retina specialists are accustomed to managing any and all complex cases involving the posterior segment. When confronting these challenging cases, we should remind ourselves of the phrase “You don’t know what you don’t know” and use the expertise of colleagues trained in ophthalmic genetics who have access to a more comprehensive suite of diagnostic tools.
TAKING THE NEXT STEP: MOLECULAR TESTING
We speak to all of our patients with IRDs about the rationale, limitations, and implications of genetic testing. This is particularly important in this era of gene-directed therapies. Thankfully, genetic testing is more accessible than ever before, with more effective tests performed at progressively lower cost. Sufficient DNA for most of our studies can be extracted from saliva kits, sparing our patients a venipuncture. Modern testing approaches demonstrate diagnostic yields nearing 70%.
The genetic testing landscape is rapidly evolving. A number of Clinical Laboratory Improvement Amendments (CLIA)–certified laboratories offer a variety of tests for patients with IRDs. Common approaches in these patients include traditional Sanger sequencing, panel-based next-generation sequencing, and whole-exome sequencing that assesses all the protein-coding regions of the genome. Stone et al advocate a clinically informed approach to focused testing of candidate genes.1 We perform focused testing in some cases and in others will direct our patients to a broader panel-testing approach.
Whatever the strategy, the ordering provider should have an understanding of the underlying technology and its limitations. Given the complexity and implications of genetic testing, genetic counselors play a central role in guiding us through the testing and counseling process.
ADDITIONAL RESOURCES FOR PATIENTS
With appropriate testing and counseling, our patients are informed and empowered to learn more about their eye condition. The following are valuable online resources:
- clinicaltrials.gov : This database of clinical studies allows patients to connect with trials specific to their disease. However, we are mindful of recent reports of blinding stem cell treatments performed under the guise of studies listed in this database.
- blindness.org : The home page of Foundation Fighting Blindness provides patients with well-vetted updates on progress in the field, informational pages about various IRDs, and opportunities to contribute and connect with the larger IRD community.
SUMMARY
Voretigene neparvovec joins the ARGUS II prosthesis as a pioneering FDA-approved intervention for IRDs. The advent of such therapies presents exciting opportunities for us and our patients but also confers new responsibilities. Retina specialists are the primary providers of eye care for many patients with IRDs. At minimum, our goal as general retina specialists is to identify cases of hereditary disease, treat secondary complications such as CME and CNV, and provide access to our patients for genetic testing. For most retinal physicians, this will ultimately involve a referral to an IRD specialist. RP
REFERENCES
- Stone EM, Andorf JL, Whitmore SS, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314-1331.
- Cideciyan AV, Swider M, Aleman TS, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007;24(5):1457-1467.
- Jain N, Jia Y, Gao SS, et al. Optical coherence tomography angiography in choroideremia: correlating choriocapillaris loss with overlying degeneration. JAMA Ophthalmol. 2016;134(6):697-702.
- Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772.
- Massof RW, Fishman GA. How strong is the evidence that nutritional supplements slow the progression of retinitis pigmentosa? Arch Ophthalmol. 2010;128(4):493-495.
- Khan KN, Mahroo OA, Islam F, Webster AR, Moore AT, Michaelides M. Functional and anatomical outcomes of choroidal neovascularization complicating BEST1-related retinopathy. Retina. 2017;37(7):1360-1370.
- Galvin JA, Fishman GA, Stone EM, Koenekoop RK. Evaluation of genotype-phenotype associations in leber congenital amaurosis. Retina. 2005;25(7):919-929.
- Pearce WA, Chen R, Jain N. Pigmentary maculopathy associated with chronic exposure to pentosan polysulfate sodium. Ophthalmology. In press.