







Photoreceptor dystrophies are a group of eye disorders that lead to visual impairment early in life. Retinitis pigmentosa (RP) is a severe inherited photoreceptor dystrophy associated with night blindness, progressive restriction of the visual fields, and eventual legal blindness.1 Approximately 10% to 20% of cases of RP are X-linked (X-linked retinitis pigmentosa, or XLRP), and roughly 70% of XLRP cases are caused by mutations in the RPGR gene.2-5 More than 20,000 people in the United States and Europe are estimated to have XLRP, and there are currently no approved treatments for patients in early stages of the disease who still have the opportunity to preserve their sight.6
CLINICAL FEATURES OF XLRP
Symptoms
XLRP is a severe inherited photoreceptor dystrophy with a median age of legal blindness of 45 years old. For perspective, this age is much younger than for other forms of RP, such as the dominant form of RP caused by mutations in the rhodopsin gene, in which the median age of legal blindness is 77 years old.7
Most patients with XLRP initially lose peripheral vision (ie, their visual field is reduced) and then progressively lose central vision (ie, decreased visual acuity). Patients with XLRP lose visual acuity at an average rate of 8.2% a year, and loss of visual acuity has been associated with thinning of the fovea.7,8
The symptoms and course of disease can vary widely among individuals with XLRP, even between those with identical disease-causing mutations. Some changes, like those in the visual field, appear to have a consistent pattern when comparing individuals with the same disease-causing mutation, but the pattern and course of other changes, such as in visual acuity, can be more variable.9
Retinal Structure and Function
XLRP is associated with the degeneration of photoreceptor cells, detected by thinning of the outer nuclear layer (ONL) on optical coherence tomography (OCT).7 Photoreceptor degeneration in XLRP is also detected by the progressive loss of rod and cone photoreceptor function on electroretinography (ERG). Most patients with RPGR-XLRP show greater loss of rod function than cone function on ERG, although approximately 20% of patients have greater or equal loss of cone function, compared to rod function.8
Based on this finding and the findings of other observational studies, there are two models of the disease course of RPGR-XLRP that will generally match most patients with the disease.8 Each model is characterized by distinct patterns of rod and cone photoreceptor degeneration across the retina (Figure 1).
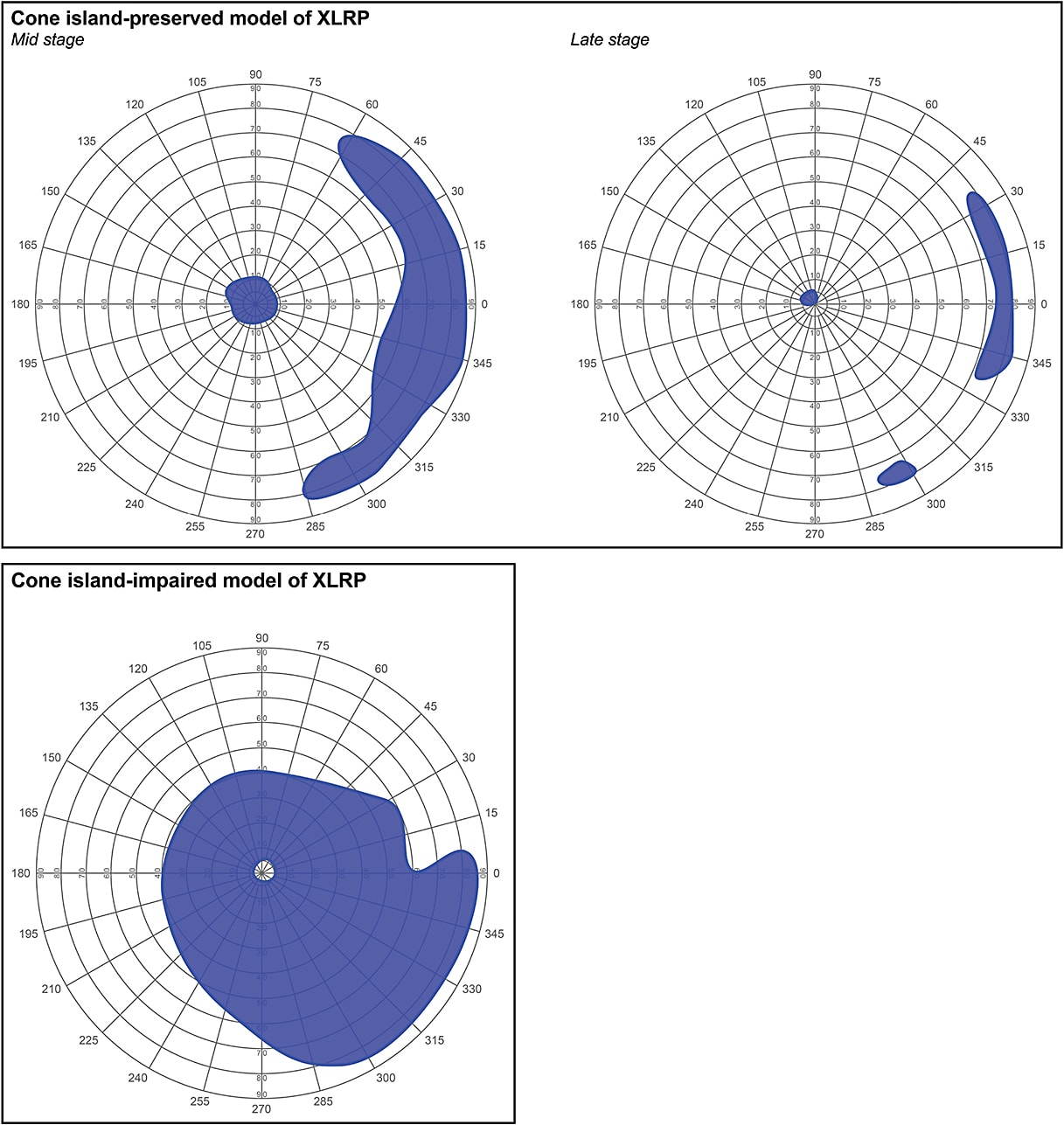
The “more common” model, known as the central island-preserved phenotype, is defined by the following stages of disease:8
- Early stage: Normal or near normal rod and cone photoreceptor function in the central island and periphery;
- Mid stage: Midperipheral scotoma separating a cone-rich central island from rod and cone photoreceptor function in the periphery; and
- Late stage: Isolated spots of residual cone function in the central island with no detectable rod or cone photoreceptor function in the periphery.
The second, “less common” model, also known as the central island-impaired phenotype, is defined by the following pattern of photoreceptor degeneration across the retina8:
- Cone photoreceptors tend to be more sensitive to impairment than rod photoreceptors;
- Loss of photoreceptors in the paracentral regions tend to isolate small spots of cone photoreceptor function in the central region; and
- Photoreceptors in the midperiphery are relatively preserved.
MOLECULAR BASIS OF XLRP
Approximately 70% of XLRP cases are caused by mutations in the RPGR gene, which encodes a GTPase regulator protein.3 The RPGR gene was initially characterized as having 19 exons with a full-length messenger RNA (mRNA) expressed in a variety of cell types (Figure 2).10
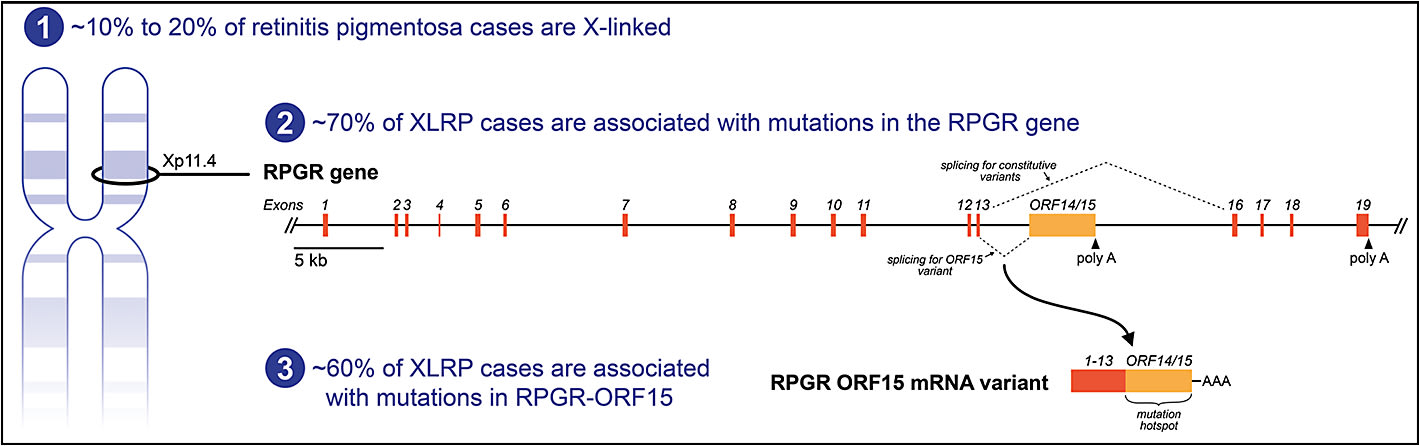
Studies have now shown, however, that alternate splicing can create multiple transcripts from the RPGR gene and that photoreceptors express an RPGR mRNA isoform that contains a unique 3’ region called ORF15.11-13 This 3’ ORF15 region is the site of up to 60% of disease-causing mutations and is therefore considered an XLRP mutation hotspot (Figure 2).13 The remaining disease-causing mutations in the RPGR gene, in addition to ORF15, are generally evenly distributed among most of the other exons (except for exons 1 and 16 through 19, which mostly lack XLRP-causing mutations).14
The RPGR gene encodes the retinitis pigmentosa GTPase regulator protein, which is located on a cilium tract that runs between the inner and outer segments of the photoreceptor. The connecting cilium is a single cylindrical column consisting of nine microtubules and anchored to the inner segment. The connecting cilium is used for the active transport of proteins between the inner and outer segments, which maintain distinct protein contents. The connecting cilium is, therefore, critical for: (1) creating a barrier between the inner and outer segments; and (2) controlling the bidirectional transport of proteins between these two discrete compartments.14,15
The exact role of RPGR protein is still under investigation, but multiple protein-binding partners have been identified, and mutations in the RPGR gene have been shown to result in abnormal protein transport across the connecting cilium, which can lead to photoreceptor cell death.14,15
ANIMAL MODELS OF XLRP
Mouse Models
Animal models of RPGR-XLRP have been essential in the preclinical evaluation of potential treatment options. Some of the models are naturally occurring genetic variants, and others have been created with the use of genetic engineering.
There are multiple mouse models of RPGR-XLRP. The naturally occurring Rd9 mouse has a short duplication in ORF15 that causes a frame shift and premature truncation of the RPGR protein. Over their lifetime, Rd9 mice have a progressive reduction in ONL thickness accompanied by progressive reductions in a-wave and b-wave amplitudes on ERG.16
To date, researchers have created three models of RPGR-XLRP by introducing null mutations into the RPGR gene:
- ∆4-6 allele: created by deleting part of exon 4 and all of exons 5 and 6;17
- ∆4: created by deleting all of exon 4;18 and
- ∆1: created by deleting all of exon 1.8
There is detectable photoreceptor loss (rods and cones) and decreased retinal function on ERG in each of the genetically engineered models, but each model is distinguished by differences in the time course and full extent of photoreceptor degeneration, the relative severity of degeneration between rod photoreceptors and cone photoreceptors, and the full extent of reduction in retina function on ERG.8,17,18
Dog Models
There are also two canine models of RPGR-XLRP, both of which were originally identified as spontaneous conditions in Siberian huskies and are called X-linked progressive retinal atrophy (XLPRA). The two forms of XLPRA are each associated with a unique mutation in ORF15 and a distinct pattern, course, and severity of the disease.19
The first form of dog XLRP, called XLPRA1, is caused by a frameshift mutation in ORF15 immediately followed by a premature stop that causes truncation of the RPGR protein. In dogs with XLPRA1, the retina develops and functions normally until approximately six months of age or later, with the first signs of disease being a decrease in the amplitude of dark-adapted rod and cone photoreceptor responses on ERG. Cone photoreceptors are typically less affected than rod photoreceptors, and the onset of end-stage retinal degeneration is usually at around 4 years old.19
The other form of dog XLRP, called XLPRA2, is caused by a nonsense mutation in ORF15. The retina develops abnormally in dogs with XLPRA2, with rod and cone photoreceptor responses on ERG at low amplitude with abnormal waveforms by five to six weeks of age. Rod and cone photoreceptors are both affected in the retinas of dogs with XLPRA2, although rod photoreceptors tend to be more severely compromised than cone photoreceptors. The midstage of the disease, with approximately 40% photoreceptor loss, occurs by 12 weeks of age, and the onset of end-stage retinal degeneration is usually at around 2 years old.19
EMERGING TREATMENTS FOR XLRP
Nutritional Supplements, CNTF, and Retinal Prosthesis
Multiple treatment approaches have been under investigation for the preservation or restoration of vision in patients with RP, with varying levels of success. Some studies have shown that various nutritional supplements, including high doses of vitamin A and docosahexaenoic acid (DHA), may slow disease progression.
Early clinical trials showed mixed results when evaluating the use of high-dose vitamin A in slowing the progression of RP. In particular, one randomized, controlled, double-masked study showed that supplementation with high-dose vitamin A (15,000 IU/day) was associated with a slower rate of decline in cone photoreceptor function (by ERG amplitude), although the study also showed that vitamin A did not help with decreases in visual field or visual acuity.20 Other research has focused on the potential benefits of combining vitamin A with additional supplements, such as lutein, with mixed success.21
DHA is an ω-3 polyunsaturated fatty acid concentrated in the membranes of photoreceptors, and it has been shown to have properties that promote photoreceptor survival. Based on these and other properties of DHA, a single-site, randomized, placebo-controlled, phase 2 clinical trial was conducted to evaluate the use of high-dose oral DHA (30 mg/kg/day) in patients with XLRP. The study showed that DHA slowed loss of the visual field over 4 years compared to placebo, but it also showed that DHA failed to slow other signs and symptoms of XLRP, including loss of visual acuity, loss of cone photoreceptor function on ERG, and thinning of the retinal outer segments on OCT.22,23
Ciliary neurotrophic factor (CNTF) is a survival factor that was shown to have a protective effect on photoreceptors in dogs with a form of RP caused by mutations in the PDE6B gene. Based on its protective effect in the PDE6B form of RP, an intravitreal injection of CNTF was evaluated in the XLPRA2 dog model of severe RPGR-XLRP. Unfortunately, CNTF was unable to prevent photoreceptor degeneration in dogs with XLPRA2 and was also associated with ocular side effects in the peripheral retina.24
The Argus II retinal prosthesis system (Second Sight Medical Products) was developed for patients with RP with little to no vision remaining. In a prospective, single-arm, unmasked, phase 2 clinical trial of patients with RP and bare-to-no light perception in both eyes, subjects had better visual function (measured by computer-based objective tests of visual skills) at 1 and 3 years after implantation of the retinal prosthesis.25 In a separate prospective, interventional case series of patients who were blind from RP or choroideremia, use of the retinal prosthesis allowed subjects to locate and grasp white objects on a black background in real three-dimensional space.26
Based on these and other studies, the Argus II retinal prosthesis was approved by the FDA in 2013 for use in patients with severe to profound RP who are 25 years old or old older, have bare light or no light perception in both eyes, and who have a previous history of vision.27 For patients with some remaining vision, however, who are in earlier vision-threatening stages of RP, gene therapy is an emerging treatment option.
Emerging Gene Therapies
There are currently several XLRP gene therapy product candidates in clinical development. Gene therapy uses engineered viruses, called viral vectors, to introduce a functional copy of a gene directly into the nuclei of a patient’s cells. When viral vectors are created, they are constructed to severely impair the ability to self-replicate.
While vectors have been made from several different types of viruses, including adenovirus and lentivirus, vectors made from adeno-associated virus (AAV) are especially well suited for delivering genes into cells of the retina because AAV is a small, simple virus amenable to genetic engineering; AAV elicits only a weak immune response after injection and has not been shown to cause diseases in humans; and vectors made from AAV have no viral genes remaining, eliminating the possibility that any viral genes will cause an adverse event.28
Preclinical Studies
The results of preclinical studies in mouse and dog models of RPGR-XLRP support the use of AAV-based gene therapies in the early to middle stages of disease. In the homozygous null ∆4-6 mouse model of RPGR-XLRP, a single subretinal injection of an AAV vector expressing either the full-length mouse or human RPGR-ORF15 gene was able to preserve retinal structure and retinal function, compared to ∆4-6 mice that received sham treatment. ∆4-6 mice that received the gene therapy were also able to maintain compartmentalized localization of intracellular proteins (ie, opsin) between the inner and outer segments, compared to mice that did not receive gene therapy.29
In another preclinical study that tested AAV-based gene therapy in both homozygous Rd9 and homozygous ∆4-6 mouse models of RPGR-XLRP, a single subretinal injection of an AAV vector expressing a codon-optimized version of the RPGR-ORF15 gene was able to preserve cone photoreceptor function under light-adapted conditions.30 A codon-optimized version of the RPGR-ORF15 gene was designed to improve sequence stability, remove cryptic splice sites, and increase expression levels after delivery of the gene into retinal cells and was confirmed to produce the full-length RPGR-ORF protein sequence with proper post-translational modifications.30
Preclinical studies in dog models have also supported the use of AAV-based gene therapies for the treatment of patients with RPGR-XLRP. In both of the naturally occurring dog models of RPGR-XLRP (XLPRA1 and XLPRA2), a single subretinal injection of an AAV vector expressing the full-length human RPGR-ORF15 gene was able to prevent photoreceptor degeneration and preserve retinal structure and function, compared to eyes that did not receive gene therapy.31 AAV-based gene therapy with the full-length RPGR-ORF15 gene was able to slow disease progression, even in dogs in the midstages of the more severe form of canine XLRP (XLPRA2).32
In another study of dogs with XLPRA2, a single subretinal injection of an AAV vector, this time expressing either a full-length stabilized or a codon-optimized human RPGR-ORF15 gene, was able to prevent photoreceptor degeneration and vision loss over a 2-year follow-up period.33 No ophthalmic alterations were seen in short-term assessments, and no detectable ocular adverse events or ONL thinning was observed in long-term assessments among dogs that received gene therapy. Rod and cone photoreceptor function (measured by ERG) and rod-mediated visual behavior (measured by maze and obstacle course testing) were improved in dogs with midstage disease that received gene therapy, compared to those that received vehicle. When comparing the retinas of dogs that received gene therapy with the full-length stabilized vs the codon-optimized version of the RPGR gene, there were no differences in ONL thickness or the intracellular localization of proteins.33
Clinical Trials
There are currently three phase 1/2 clinical trials evaluating the safety and therapeutic potential of RPGR gene therapy in patients with RPGR-XLRP.
Nightstar Therapeutics is conducting a nonrandomized, open-label, multicenter, dose-escalation trial of a single subretinal injection of an AAV vector expressing the human RPGR gene in patients with RPGR-XLRP (NCT03116113). The study is testing three dose levels (low, medium, and high) and enrolling up to 24 subjects. The primary endpoint is safety at 1 year after injection, which includes an assessment of the proportions of patients with dose-limiting toxicity and with treatment-emergent adverse events. Secondary endpoints include measures of potential effectiveness, such as change from baseline in visual acuity.
MeiraGTx is also conducting a nonrandomized, open-label, multicenter, dose-escalation trial of a single subretinal injection of an AAV vector expressing the human RPGR gene in patients with RPGR-XLRP (NCT03252847). This study is also testing three dose levels (low, medium, and high) but is enrolling up to 36 subjects. The primary endpoint is safety at 18 months after injection, in part assessed by the incidence of adverse events related to the subretinal injection of AAV-RPGR. Secondary endpoints include changes in visual function and quality of life.
Applied Genetic Technologies Corporation (AGTC) is conducting the third study, a nonrandomized, open-label, multicenter, dose-escalation trial of a single subretinal injection of an AAV vector expressing the human RPGR gene in patients with XLRP caused by mutations in RPGR-ORF15 (NCT03316560). Up to 15 subjects are being enrolled in the study, and one eye from each subject will receive a single subretinal injection of an AAV vector expressing a full-length stabilized version of the human RPGR gene.
The study design is divided into two stages. The first stage is testing three dose levels, beginning with the lowest dose and proceeding to a higher dose only after review of safety data by a Data and Safety Monitoring Committee. The second stage of the study will test the maximum tolerated dose (as determined from the first stage) in a separate subject cohort. Subjects in the first stage are at least 18 years of age, and subjects in the second stage are at least 6 years of age. The primary endpoint is safety at 1 year after injection and will include an assessment of ocular and nonocular adverse events and hematology and clinical chemistry parameters. Select secondary endpoints include measures of visual acuity (with the ETDRS chart), visual field (with static perimetry), and retinal structure (with OCT).
SUMMARY
Approximately 20,000 people in the United States and Europe are estimated to have XLRP, an inherited retinal disease that causes progressive vision loss and eventual blindness. Multiple treatment approaches have been under investigation for the preservation or restoration of vision in patients with RP, with varying levels of success.
There have been mixed results with the use of nutritional supplements, such as vitamin A, while an intravitreal injection of CNTF did not prevent photoreceptor degeneration in a dog model of RPGR-XLRP. The Argus II retinal prosthesis can bring limited sight back to patients who have lost most to all of their vision, but there remains no approved treatment for patients at earlier stages of progressive RP who still have the opportunity to preserve their sight.
Preclinical studies have shown promising results for AAV-based gene therapies in the preservation of the retinal structure and function in mouse and dog models of RPGR-XLRP with early and midstage RP. The results of these studies have supported the advancement of XLRP gene therapy product candidates into clinical development, with initial results from the first phase 1/2 clinical trials expected in 2019. RP
REFERENCES
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132-141.
- Bird AC. X-linked retinitis pigmentosa. Br J Ophthalmol. 1975;59:177-199.
- Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545-1554.
- Fishman GA. Retinitis pigmentosa. Genetic percentages. Arch Ophthalmol. 1978;96:822-826.
- Haim M. Prevalence of retinitis pigmentosa and allied disorders in Denmark. III. Hereditary pattern. Acta Ophthalmol (Copenh). 1992;70:615-624.
- Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151-158.
- Sandberg MA, Rosner B, Weigel-DiFranco C, Dryja TP, Berson EL. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Invest Ophthalmol Vis Sci. 2007;48:1298-1304.
- Huang WC, Wright AF, Roman AJ, et al. RPGR-associated retinal degeneration in human X-linked RP and a murine model. Invest Ophthalmol Vis Sci. 2012;53:5594-5608.
- Zahid S, Khan N, Branham K, et al. Phenotypic conservation in patients with X-linked retinitis pigmentosa caused by RPGR mutations. JAMA Ophthalmol. 2013;131:1016-1025.
- Meindl A, Dry K, Herrmann K, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13:35-42.
- Hong DH, Li T. Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Invest Ophthalmol Vis Sci. 2002;43:3373-3382.
- Hong DH, Pawlyk B, Sokolov M, et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413-2421.
- Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462-6.
- Hosch J, Lorenz B, Stieger K. RPGR: role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. 2011;32:1-11.
- Megaw RD, Soares DC, Wright AF. RPGR: its role in photoreceptor physiology, human disease, and future therapies. Exp Eye Res. 2015;138:32-41.
- Thompson DA, Khan NW, Othman MI, et al. Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS One. 2012;7:e35865.
- Hong DH, Pawlyk BS, Shang J, et al. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A. 2000;97:3649-3654.
- Brunner S, Skosyrski S, Kirschner-Schwabe R, et al. Cone versus rod disease in a mutant Rpgr mouse caused by different genetic backgrounds. Invest Ophthalmol Vis Sci. 2010;51:1106-1115.
- Zhang Q, Acland GM, Wu WX, et al. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002;11:993-1003.
- Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111:761-772.
- Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128:403-411.
- Hoffman DR, Hughbanks-Wheaton DK, Pearson NS, et al. Four-year placebo-controlled trial of docosahexaenoic acid in X-linked retinitis pigmentosa (DHAX trial): a randomized clinical trial. JAMA Ophthalmol. 2014;132:866-873.
- Hoffman DR, Hughbanks-Wheaton DK, Spencer R, et al. Docosahexaenoic acid slows visual field progression in X-linked retinitis pigmentosa: ancillary outcomes of the DHAX trial. Invest Ophthalmol Vis Sci. 2015;56:6646-6653.
- Beltran WA, Wen R, Acland GM, Aguirre GD. Intravitreal injection of ciliary neurotrophic factor (CNTF) causes peripheral remodeling and does not prevent photoreceptor loss in canine RPGR mutant retina. Exp Eye Res. 2007;84:753-771.
- Ho AC, Humayun MS, Dorn JD, et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology. 2015;122:1547-1554.
- Luo YH, Zhong JJ, da Cruz L. The use of Argus(R) II retinal prosthesis by blind subjects to achieve localisation and prehension of objects in 3-dimensional space. Graefes Arch Clin Exp Ophthalmol. 2015;253:1907-1914.
- Second Sight Medical Products receives FDA approval for Argus II system [news release]. Sylmar, CA: Second Sight Investor Relations; February 14, 2013. http://investors.secondsight.com/releasedetail.cfm?ReleaseID=873595 . Accessed October 25, 2017.
- Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317-334.
- Wu Z, Hiriyanna S, Qian H, et al. A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum Mol Genet. 2015;24:3956-3970.
- Fischer MD, McClements ME, Martinez-Fernandez de la Camara C, et al. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol Ther. 2017;25:1854-1865.
- Beltran WA, Cideciyan AV, Lewin AS, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109:2132-2137.
- Beltran WA, Cideciyan AV, Iwabe S, et al. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc Natl Acad Sci U S A. 2015;112:E5844-E5853.
- Beltran WA, Cideciyan AV, Boye SE, et al. Optimization of retinal gene therapy for X-linked retinitis pigmentosa due to RPGR mutations. Mol Ther. 2017;25:1866-1880.