







Emerging Treatment for X-linked Retinoschisis
A rare inherited retinal disease with no current treatment meets ocular gene therapy.
PAUL YANG, MD, PhD
X-linked retinoschisis (XLRS) is an orphan inherited disease of the retina caused by mutations in the RS1 gene, which encodes the retinoschisin protein.1 Retinoschisin is a structural protein expressed and secreted by photoreceptor and bipolar cells that binds strongly and specifically to the surfaces of many cells in the retina.2-4
Mutated forms of retinoschisin are unable to bind properly, which leads to schisis, or splitting of layers of the retina. The incidence of XLRS is estimated to be between 1 in 5,000 and 1 in 20,000 males, with an estimated prevalence of 35,000 in the United States and Europe combined.5,6 There are currently no approved treatments for patients with XLRS.
CLINICAL FEATURES OF X-LINKED RETINOSCHISIS
X-linked retinoschisis is thought to be completely penetrant among individuals with mutations in the RS1 gene; however, the expression of the phenotype is highly variable with regard to the severity of the retinoschisis and central vision loss, even among affected members of the same family.
Classically, patients with XLRS present during school age with foveal retinoschisis and bilaterally decreased visual acuity in the range of 20/60 to 20/120.7-10 In some cases, XLRS is diagnosed much earlier in infants due to the presence of related complications, such as retinal detachment or vitreous hemorrhage.11,12 Although XLRS is bilateral, there can be asymmetric secondary complications that lead to unequal vision loss between eyes.9,13
Paul Yang, MD, PhD, is assistant professor of ophthalmology at the Casey Eye Institute, Oregon Health & Science University in Portland. He reports no financial interests in products mentioned in this article. Dr. Yang can be reached via e-mail at yangp@ohsu.edu.
Almost all XLRS patients have maculoschisis, or splitting of the layers of the macula, and diagnosis is often made based on a characteristic spoke-wheel pattern appearance of the macula on fundus ophthalmoscopy (Figure 1).7-10,14,15 In some cases, maculoschisis may cause the macula to have a honeycomb appearance and there may be evidence of hypopigmentation.14
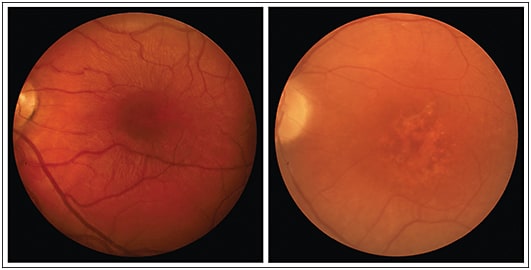
Figure 1. X-linked retinoschisis (XLRS) as seen by fundus ophthalmoscopy. The left fundus photograph is from a 10-year-old XLRS patient with classic maculoschisis showing the characteristic spoke-wheel pattern appearance. The right fundus photograph is from a 33-year-old XLRS patient with thinned, or “collapsed,” atrophic macular morphology.
RIGHT IMAGE COURTESY OF DR. MARK PENNESI, CASEY EYE INSTITUTE, OREGON HEALTH AND SCIENCE UNIVERSITY.
Imaging Studies
Images captured by optical coherence tomography confirm splitting and disorganization of the retinal layers in patients with XLRS (Figure 2, page 66).16 Retinal thickness is often increased by the presence of schisis cavities, and OCT studies have revealed that splitting can occur within the multiple layers that constitute the retina, including the nerve fiber layer, inner nuclear layer, outer nuclear layer, and outer plexiform layer.
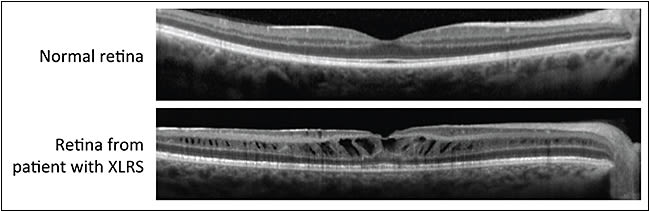
Figure 2. X-linked retinoschisis (XLRS) as seen by optical coherence tomography (OCT). The top OCT scan is through the fovea of a normal retina. The bottom OCT scan is through the retina of a patient with XLRS.
COURTESY OF DR. MARK PENNESI, CASEY EYE INSTITUTE, OREGON HEALTH AND SCIENCE UNIVERSITY.
Maculoschisis can be centered over the fovea or restricted to the parafoveal region.15 While almost all patients have maculoschisis, only approximately half of patients also have peripheral retinoschisis.7-10
Approximately half of XLRS patients have a characteristic abnormality on full-field electroretinogram (ERG) testing, with the response to a bright flash of light under scotopic conditions producing a negative waveform in which the b-wave to a-wave amplitude ratio is less than one.10,17
It was suggested in one study that the nature and severity of the negative waveform ERG response in XLRS is correlated with patient age and the putative severity of the disease-causing mutation in the RS1 gene.18
The course of XLRS disease progression is under active investigation. Some studies have suggested that macular pathology and visual symptoms do not progress significantly beyond what are found at initial diagnosis.5
Other studies have identified significant age-related associations, which indicate that younger XLRS patients are more likely to have thicker retinas with intraretinal schisis cavities, while older patients are more likely to have thinner retinas with a flattened, or “collapsed,” macular morphology.19,20
Two natural history studies are currently in progress to characterize disease progression in patients with XLRS. The National Eye Institute is conducting one trial (NCT00055029), and Applied Genetic Technologies Corporation (AGTC; Alachua, FL) is conducting the other (NCT02331173).21,22
MOLECULAR FEATURES OF X-LINKED RETINOSCHISIS
The RS1 gene is 32.4 kb in length and located on the short arm of the X chromosome (Figure 3, page 67). The RS1 gene encodes the retinoschisin peptide, which is 24 kDa, contains 224 amino acids, and is assembled into a homo-octamer before secretion into the extracellular space.1,23,24
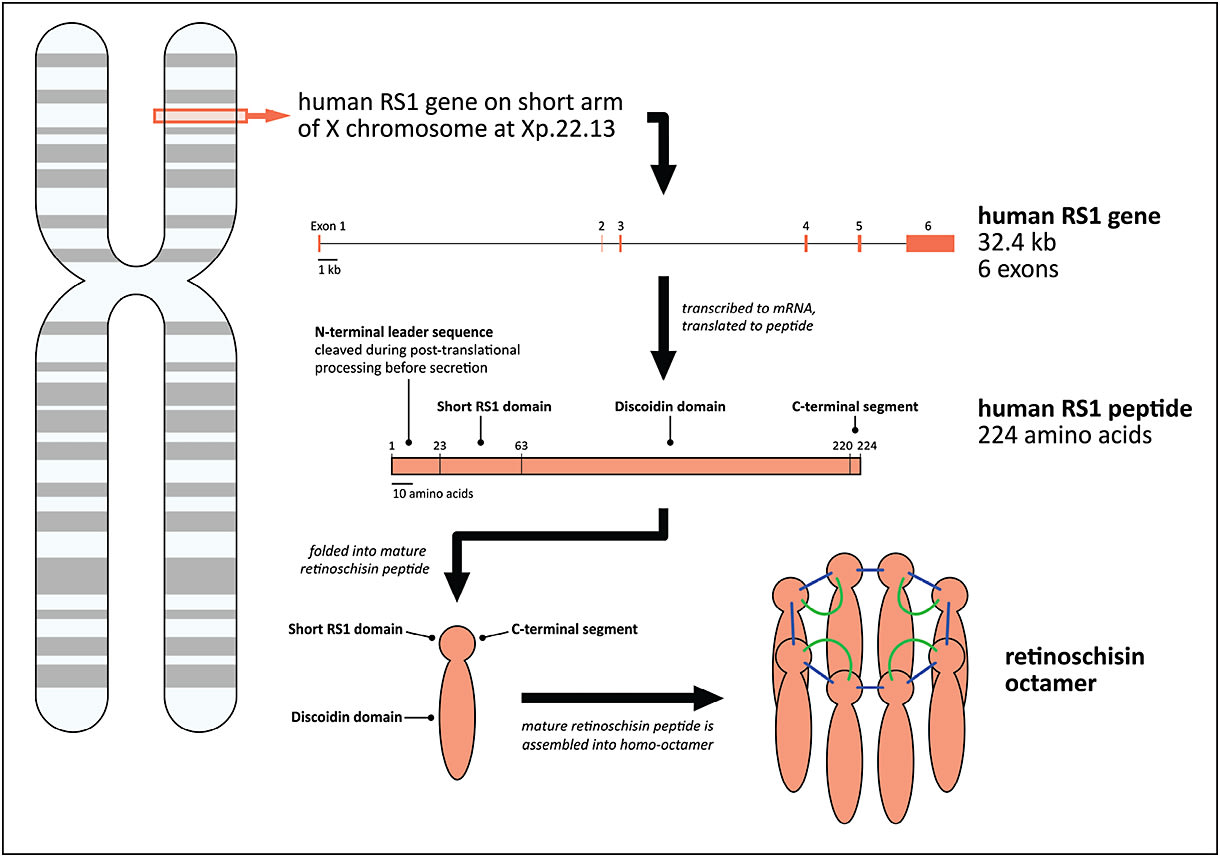
Figure 3. Molecular features of retinoschisin (RS1). Schematic representations of folded retinoschisin peptide and retinoschisin octamer modified from Wu et al27 and Molday et al.5
COURTESY OF ELSEVIER.
Up to 40% of disease-causing RS1-associated variants result in nonsense or frameshift mutations, which prevent the production of the full-length retinoschisin peptide. Any truncated peptide that may be produced is usually unstable and quickly degraded by the cell. In contrast, more than 50% of disease-causing RS1 mutations are missense alterations that are compatible with the production of a full-length, but abnormal, peptide.5
The retinoschisin peptide is composed of an N-terminal leader sequence, RS1 domain, discoidin domain, and C-terminal segment, each of which has been found to contain disease-causing mutations in XLRS patients.
N-terminal Leader Sequence
This sequence is critical for trafficking freshly translated retinoschisin to sites of post-translational processing, and it is cleaved during the peptide maturation process before secretion.
Disease-causing mutations in the N-terminal leader sequence can lead to the production of abnormal retinoschisin peptides, which are mislocalized during post-translational processing. As a result, the cell will degrade mislocalized retinoschisin, ultimately leading to an extracellular deficiency.25-27
Short RS1 Domain
This structural domain is critical for the intracellular assembly of RS1 peptides into a homo-octameric complex. Disease-causing mutations in the short RS1 domain lead to the production of an abnormal retinoschisin peptide that does not form a homo-octamer. Post-translational processing and secretion appear to be unaffected, emphasizing the vital role of octamer formation in retinoschisin function in the retina.24,27
Discoidin Domain
This evolutionarily conserved structural domain is found in a variety of prokaryotic and eukaryotic transmembrane and extracellular proteins, and it is associated with cell adhesion, migration, and signaling.5
Disease-causing mutations in the RS1 discoidin domain are associated with abnormal folding of the retinoschisin peptide and the formation of disordered retinoschisin aggregates that cannot be secreted from the cell.25-27
C-terminal Segment
This segment is critical for the assembly of RS1 peptides into a homo-octameric complex. Similar to the short RS1 domain, disease-causing mutations in the C-terminal segment can lead to the production of abnormal retinoschisin peptides that are properly folded and secreted but that cannot be assembled into an octamer.24,27
EMERGING OPTIONS FOR TREATMENT
There are currently no approved treatment options for patients with XLRS. The management of decreased VA in XLRS is limited to the use of low-vision aids, such as large-text or high-contrast reading materials.5
Some reports have suggested that topical carbonic anhydrase inhibitors, such as dorzolamide (Trusopt, Merck, Kenilworth, NJ), may provide some reduction in the degree of schisis; however, most of these reports are anecdotal or based on retrospective case studies.28,29
One prospective, randomized clinical trial reported that best corrected VA was improved by 0.1 logMAR (5 ETDRS letters) in six of 11 patients after three months of treatment with oral acetazolamide, but there was no correlation with changes in the degree of schisis.30
Gene Therapies
Gene therapy is an emerging treatment option for patients with XLRS, and product candidates based on adeno-associated viral (AAV) vectors expressing the human RS1 gene are currently in clinical development.
Gene therapy involves the introduction of a functional copy of a gene into a patient’s own cells using a delivery system, most commonly based on a viral vector, to treat a genetic defect. Gene therapy can provide restorative disease-modifying effects by correcting the underlying cause of the disease.
Adeno-associated virus is a small, simple, nonenveloped, nonpathogenic virus that elicits only a weak immune response and has never been linked to human disease, making it particularly well suited for gene therapy-based treatment of retinal diseases. AAV vectors have been optimized for gene therapy by removing the two native viral genes and replacing them with a human gene encoding a therapeutic protein.
Because AAV vectors have no viral genes remaining, the possibility that any viral genes will cause an adverse event is virtually eliminated.31
Preclinical studies have been conducted to assess the therapeutic potential of gene therapy-based approaches for the treatment of XLRS and, more specifically, to investigate the potential safety and effectiveness of AAV vectors expressing RS1 in animal models.
In a mouse model of XLRS, subretinal or intravitreal injection of AAV vectors expressing mouse or human RS1 improved the organization of the retinal cell layers and restored ERG b-wave responses under scotopic conditions.32,33
Safety studies in large mammals including nonhuman primates, as well as rabbits, showed that an intravitreal injection of AAV vectors expressing human RS1 is safe but can be associated with dose-related, self-limited anterior- and posterior-segment inflammation that improves over time.34,35
Clinical Trials of AAV
The results of these and other preclinical studies supported the advancement of gene therapy-based XLRS product candidates into clinical development, and two phase 1/2 trials are currently being conducted to evaluate the safety and tolerability of an intravitreal injection of AAV vector expressing human RS1 in patients with XLRS.
The NEI is conducting a single-center nonrandomized, open-label, dose-escalation trial (NCT02317887).36 Participants are males with XLRS who are 18 years of age or older. One eye of each participant is receiving a single intravitreal injection of AAV expressing human RS1. The study design is divided into two stages, which will together enroll a total of up to 15 participants.
In the first stage, initial participants are receiving low doses, and subsequent participants will receive higher doses. In the second stage, new participants will be enrolled to receive a dose that was identified as well tolerated and potentially efficacious during the first stage.
Safety assessments include an analysis of adverse events, laboratory test results, retinal function, and ocular structure. Secondary outcomes include changes in visual function after injection, ERG responses, changes in retinal structure as assessed by OCT, visual field measurements, and immune response to AAV and RS1.36
AGTC is conducting a multicenter, nonrandomized, open-label, dose-escalation trial (NCT02416622) at four study sites in the United States.37 One eye of each participant is receiving a single intravitreal injection of AAV expressing human RS1. The AGTC XLRS phase 1/2 study is divided into two stages, which will together enroll a total of up to 27 participants.
In the first stage, three dose levels are being evaluated, beginning with the lowest dose level and proceeding to higher dose levels after review of safety data by a Data and Safety Monitoring Committee. Participants in this initial dose-escalation stage are males with XLRS who are 18 years of age or older.
In the second stage, additional participants will be enrolled to evaluate the maximum tolerated dose that was determined during the first stage. Participants in the second stage will be males with XLRS who are six years of age or older.
The safety assessments include an analysis of the number and proportion of participants experiencing adverse events and immune response to RS1. Efficacy assessments include changes in VA and schisis cavity size after injection.37
FUTURE DIRECTIONS
X-linked retinoschisis is a rare inherited retinal disease for which there is currently no approved treatment. The management of decreased VA in XLRS is limited to the use of low-vision aids, such as large-text or high-contrast reading materials.
Organizations and companies that are committed to restoring sight to individuals with rare genetic eye disorders, including the NEI and AGTC, currently have advanced AAV-based gene therapy product candidates in clinical development, and they are leading the future of XLRS treatment. Preliminary results from these phase 1/2 trials are expected late 2015 to early 2016. RP
REFERENCES
1. Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17:164-170.
2. Molday LL, Hicks D, Sauer CG, Weber BH, Molday RS. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci. 2001;42:816-825.
3. Reid SN, Akhmedov NB, Piriev NI, et al. The mouse X-linked juvenile retinoschisis cDNA: expression in photoreceptors. Gene. 1999;227:257-266.
4. Takada Y, Fariss RN, Tanikawa A, et al. A retinal neuronal developmental wave of retinoschisin expression begins in ganglion cells during layer formation. Invest Ophthalmol Vis Sci. 2004;45:3302-3312.
5. Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31:195-212.
6. Sikkink SK, Biswas S, Parry NR, Stanga PE, Trump D. X-linked retinoschisis: an update. J Med Genet. 2007;44:225-232.
7. George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol. 1996;114:274-280.
8. Pimenides D, George ND, Yates JR, et al. X-linked retinoschisis: clinical phenotype and RS1 genotype in 86 UK patients. J Med Genet. 2005;42:e35.
9. Roesch MT, Ewing CC, Gibson AE, Weber BH. The natural history of X-linked retinoschisis. Can J Ophthalmol. 1998;33:149-158.
10. Wang NK, Liu L, Chen HM, et al. Clinical presentations of X-linked retinoschisis in Taiwanese patients confirmed with genetic sequencing. Mol Vis. 2015;21:487-501.
11. Tantri A, Vrabec TR, Cu-Unjieng A, et al. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49:214-230.
12. Lee JJ, Kim JH, Kim SY, Park SS, Yu YS. Infantile vitreous hemorrhage as the initial presentation of X-linked juvenile retinoschisis. Korean J Ophthalmol. 2009;23:118-120.
13. Tanino T, Katsumi O, Hirose T. Electrophysiological similarities between two eyes with X-linked recessive retinoschisis. Doc Ophthalmol. 1985;60:149-161.
14. Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina. 2005;25:612-618.
15. Yu J, Ni Y, Keane PA, et al. Foveomacular schisis in juvenile X-linked retinoschisis: an optical coherence tomography study. Am J Ophthalmol. 2010;149:973-978 e972.
16. Stanga PE, Chong NH, Reck AC, Hardcastle AJ, Holder GE. Optical coherence tomography and electrophysiology in X-linked juvenile retinoschisis associated with a novel mutation in the XLRS1 gene. Retina. 2001;21:78-80.
17. Renner AB, Kellner U, Fiebig B, et al. ERG variability in X-linked congenital retinoschisis patients with mutations in the RS1 gene and the diagnostic importance of fundus autofluorescence and OCT. Doc Ophthalmol. 2008;116:97-109.
18. Bowles K, Cukras C, Turriff A, et al. X-linked retinoschisis: RS1 mutation severity and age affect the ERG phenotype in a cohort of 68 affected male subjects. Invest Ophthalmol Vis Sci. 2011;52:9250-9256.
19. Lesch B, Szabo V, Kanya M, et al. Clinical and genetic findings in Hungarian patients with X-linked juvenile retinoschisis. Mol Vis. 2008;14:2321-2332.
20. Menke MN, Feke GT, Hirose T. Effect of aging on macular features of X-linked retinoschisis assessed with optical coherence tomography. Retina. 2011;31:1186-1192.
21. Clinical evaluation of patients with X-linked retinoschisis. Clinicaltrials.gov Web site. Available at: https://clinicaltrials.gov/ct2/show/NCT02331173?term=XLRS&rank=1. Accessed July 21, 2015.
22. Clinical and genetic studies of X-linked juvenile retinoschisis. Clinicaltrials.gov Web site. Available at: https://clinicaltrials.gov/ct2/show/NCT00055029?term=XLRS&rank=3. Accessed July 21, 2015.
23. Molday RS. Focus on molecules: retinoschisin (RS1). Exp Eye Res. 2007;84:227-228.
24. Wu WW, Wong JP, Kast J, Molday RS. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. 2005;280:10721-10730.
25. Vijayasarathy C, Sui R, Zeng Y, et al. Molecular mechanisms leading to null-protein product from retinoschisin (RS1) signal-sequence mutants in X-linked retinoschisis (XLRS) disease. Hum Mutat. 2010;31:1251-1260.
26. Wang T, Waters CT, Rothman AM, et al. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum Mol Genet. 2002;11:3097-3105.
27. Wu WW, Molday RS. Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X-linked retinoschisis. J Biol Chem. 2003;278:28139-28146.
28. Apushkin MA, Fishman GA. Use of dorzolamide for patients with X-linked retinoschisis. Retina. 2006;26:741-745.
29. Genead MA, Fishman GA, Walia S. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with X-linked retinoschisis. Arch Ophthalmol. 2010;128:190-197.
30. Gurbaxani A, Wei M, Succar T, et al. Acetazolamide in retinoschisis: a prospective study. Ophthalmology. 2014;121:802-803 e803.
31. Goncalves MA. Adeno-associated virus: from defective virus to effective vector. Virol J. 2005;2:43.
32. Min SH, Molday LL, Seeliger MW, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of X-linked juvenile retinoschisis. Mol Ther. 2005;12:644-651.
33. Park TK, Wu Z, Kjellstrom S, et al. Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther. 2009;16:916-926.
34. Marangoni D, Wu Z, Wiley HE, et al. Preclinical safety evaluation of a recombinant AAV8 vector for X-linked retinoschisis after intravitreal administration in rabbits. Hum Gene Ther Clin Dev. 2014;25:202-211.
35. Ye GJ, Budzynski E, Sonnentag P, et al. Safety and biodistribution evaluation in cynomolgus macaques of rAAV2tYF-CB-hRS1, a recombinant adeno-associated virus vector expressing retinoschisin. Hum Gene Ther Clin Dev. 2015;26:165-176.
36. Study of RS1 ocular gene transfer for X-linked retinoschisis. Clinicaltrials.gov Web site. Available at: https://clinicaltrials.gov/ct2/show/NCT02317887?term=NCT02317887&rank=1. Accessed July 19, 2015.
37. Safety and efficacy of rAAV-hRS1 in patients with X-linked retinoschisis (XLRS). Clinicaltrials.gov Web site. Available at: https://clinicaltrials.gov/ct2/show/NCT02416622?term=NCT02416622&rank=1. Accessed July 19, 2015.