







PEER REVIEWED
Genetic Therapies for Inherited Retinal Disease
Adeno-associated viruses have shown promise.
SANFORD L. BOYE, MS • SHANNON E. BOYE, PhD
When people are asked which of their five senses they would least prefer to lose, the overwhelming majority answer, “my sight.” Recently, successes in the field of gene therapy have offered hope to patients with inherited retinal disease that their vision may be preserved or even restored.
While other gene therapy vectors are being tested, this article will summarize adeno-associated virus (AAV)–mediated gene replacement strategies currently being tested in clinical trials and those demonstrating potential for clinical application in the near future, and it will conclude with a discussion of new directions being pursued in the field with the goal of expanding the use of AAV-mediated gene therapy to treat retinal disease.
AAV: THE GOLD STANDARD IN RETINAL GENE THERAPY
In clinical trials, AAV has proved to be safe and, of the viral vectors available for gene delivery to the retina, the most effective.1 In terms of preclinical development, it is also preferred over other platforms due to the wide variety of capsid variants available, which allow for selective targeting of different retinal cells, ease of use (relatively facile manipulation of genetic elements), the ability to drive persistent transgene expression, and low immunogenicity.2
In the late 1990s, preliminary work proved that AAV could deliver genes encoding reporter proteins such as GFP to the retina. In 2001, the first convincing example of therapeutic gene delivery to a large animal was reported in a dog model of RPE65 Leber congenital amaurosis 2 (LCA2).3 Gene replacement in this model led to lifelong restoration of vision in treated dogs.
Sanford L. Boye, MS, is associate scientist, and Shannon E. Boye, PhD, is assistant professor and head of the Shannon E. Boye Laboratory at the University of Florida College of Medicine in Gainesville. Both authors are among the holders of US patent #61/327,521, a recombinant adeno-associated viral vector. Dr. Boye can be at Shannon.Boye@eye.ufl.edu.
Since then, AAV-mediated gene replacement has been used to restore vision in a multitude of inherited retinal disease models.1 Its utility is best exemplified by AAV thus far being the only viral vector to show efficacy in clinical trials for treating inherited retinal disease.4-13
DISEASES TREATED WITH AAV GENE THERAPY
RPE65 Leber Congenital Amaurosis 2
Mutations in RPE65 (OMIM #180069) are associated with a form of Leber congenital amaurosis, a severe early onset retinal disease. RPE65 (retinal pigment epithelium-specific 65 kDa protein) encodes a protein expressed in RPE cells that helps to supply chromophore (11 cis-retinal) to photoreceptors, thereby allowing them to respond to light.
Patients with LCA2 exhibit profound dysfunction and progressive degeneration of photoreceptors. When delivered subretinally by AAV2 vector, RPE65 supplementation has restored rod and cone function and visually guided behavior in multiple animal models of LCA2, including the RPE65 mutant dog, the Rpe65-/- mouse, and the naturally occurring RPE65 mutant, the rd12 mouse.14 These studies were extended to evaluate the efficiency of alternate AAV serotypes, self-complimentary genomes, and windows of therapy.
Good Laboratory Practice safety studies were conducted by investigators at the University of Florida and University of Pennsylvania to ensure the safety and efficacy of clinical vector (AAV2-CBA-RPE65) in nonhuman primates (NHPs) and rd12 mice, respectively.15,16 The results of these studies supported the initiation of multiple phase 1/2 gene therapy trials involving unilateral, subretinal injections of AAV-RPE65 at University College London, the University of Pennsylvania, and the University of Florida.
A recounting of the timelines and differences in philosophies of the competing groups was presented in a 2008 article in Science.17 The short-term (one to three years) results of these trials have now been reported.4-13 Despite small differences in vector choices and clinical trial designs, all of the trials reported efficacy (quantification of increased light sensitivity and demonstration of improved useful vision) in the absence of any vector-related adverse events.
Given this success, the decision was made to administer treatment to the second eye of previously treated patients (1.7-3.3 years later), which proved equally safe and effective.18
Phase 3 clinical trials employing bilateral gene replacement (with treatments separated by a matter of days) were initiated in 2013 by investigators at the Children’s Hospital of Philadelphia (CHOP) and the University of Iowa and by Spark Therapeutics (Philadelphia, PA), a gene therapy–based company emanating from CHOP. The results of these trials are anticipated later this year.
Spark announced in late 2014 that the FDA had granted “breakthrough” status to its exclusively licensed AAV-RPE65 (SPK-RPE65), which the company hopes will expedite approval of this therapy for broad use.
In the meantime, long-term follow up continues on LCA2 patients treated in phase 1/2 trials by the University of Pennsylvania/University of Florida/University College London teams. They reported that, despite enduring improvements in retinal function, treated patients exhibited unabated photoreceptor degeneration.8 This report suggested the need for further refinement of the gene replacement strategy. Focus is now being placed on evaluating persistence of functional gains in these patients.
Choroideremia
Mutations in CHM (OMIM #300390) are associated with choroideremia, an X-linked inherited retinal disease. CHM encodes Rab escort protein -1 (REP1), which participates in the prenylation of proteins and is thought to be involved in the trafficking of proteins in both the RPE and photoreceptors. Affected men and boys exhibit loss of the RPE/choriocapillaris, photoreceptor degeneration, retinal remodeling, and profound loss of vision within three decades. Female carriers may exhibit pigmentary fundus changes.
Work in mouse models has suggested that, while CHM mutations affect both the RPE and photoreceptors in a cell-autonomous fashion, RPE defects accelerate photoreceptor degeneration.19,20 Unlike LCA2, a large animal model does not exist, and mouse models less faithfully mimic the CHM patient phenotype, making preclinical proof-of-concept work more challenging.
The results of experiments using different viral vectors (Adenovirus and Lentivirus) to deliver CHM to either patient cells ex vivo or to Chm-deficient mice, respectively, were used to support a phase 1/2 clinical trial to treat patients using a subretinally delivered AAV vector carrying REP1 (ClinicalTrials.gov NCT01461213).21-23
The serotype (AAV2) and promoter (CBA) were chosen for their proven ability to drive transgene expression in both the RPE and photoreceptors and their proven track record in LCA2 trials.
The results of a multicenter clinical trial involving six affected men revealed increases in mean retinal sensitivity (five of six patients) and improvements in visual acuity (two of six patients).23 Fluctuations in untreated control eyes were also observed. A phase 1/2 clinical trial utilizing AAV2 was initiated by Spark in January 2015 (NCT02341807).
More recently, improved efficacy has been shown in a mouse model of CHM using AAV8, as well as in induced pluripotent stem cell–derived RPE cells from CHM patients using AAV5.24.25 Investigators in both studies have suggested that AAV8 and AAV5 vectors may offer advantages over AAV2 for treating CHM.
Autosomal-recessive Retinitis Pigmentosa
Mutations in mer receptor tyrosine kinase (MERTK; OMIM #604705) are associated with a rare form of autosomal recessive retinitis pigmentosa (RP).26 MERTK encodes a protein by the same name, which participates in the phagocytosis of photoreceptor outer segments by the RPE. In its absence, debris accumulates in the subretinal space, leading ultimately to photoreceptor degeneration and vision loss.
One of the most famous animal models of inherited retinal disease, the Royal College of Surgeons rat, carries a mutation in MERTK, and this model has been used to test the efficacy of gene replacement with various viral vectors.27-31 Preclinical evaluation of an AAV2 vector carrying an RPE-specific promoter to drive expression of MERTK showed it to be safe and efficacious.31 These findings supported the initiation of a phase 1 clinical trial in Saudi Arabia at the King Faisal Specialist Hospital and Research Center (NCT01482195). Six patients have now been treated, and the results of their two-year follow up will be published soon (Alkuraya F, Abboud E, personal communication, 2014).
Leber Hereditary Optic Neuropathy
Mutations in mitochondrial genes, such as nicotinamide-adenine dinucleotide dehydrogenase subunit 4 (ND4; OMIM #516003), are associated with Leber hereditary optic neuropathy (LHON), a maternally inherited disease affecting ganglion cells in the inner retina and causing blindness in the second/third decade of life.32
Rodent models of ND4 LHON have been created by delivering mutant human ND4 cDNA, containing cytoplasmic rather than mitochondrial triplet code and a signal sequence for transport into mitochondria, or simply via electroporation of mutant ND4.33,34
These models have recapitulated the ganglion cell degeneration, optic nerve atrophy, and visual loss seen in LHON patients. Delivery of wild-type ND4 to the mitochondria of ganglion cells, using AAV vectors employing the same “allotopic expression” strategy, have rescued phenotype.35,36
Taken together, these studies have supported the initiation of multiple phase 1/2 clinical trials led by the University of Miami (NCT02161380), GenSight Biologics (Paris, France; NCT02064569), and Huazhong University of Science and Technology in China (NCT01267422), all of which are actively recruiting.
Interestingly, phenotype in the animal model could also be rescued by directly infecting the mitochondria with an AAV vector,37 which was accomplished by incorporating a mitochondrial targeting sequence on the AAV capsid that then delivered the mitochondrial coded gene.
Wet AMD
VEGF plays a significant role in the neovascularization associated with the exudative (wet) form of age-related macular degeneration. Current treatment involves repeated intravitreal injections of VEGF antibodies, RNA aptamers, or soluble receptors. While effective, the need for repeated (monthly) injections poses a burden to patients.
Subretinal injection of AAV-carrying soluble FLT-1 (sFLT1; Avalanche Biotechnologies, Menlo Park, CA), a portion of the R1 VEGF receptor, promotes the binding of VEGF, thereby blocking binding of its intended receptors on the vascular endothelium and inhibiting neovascularization in animal models.38 This preliminary work, spearheaded by investigators at the University of Western Australia, formed the basis for phase 1/2 clinical trials led by Avalanche (NCT01494805).
The University of Florida and Genzyme (Cambridge, MA) have investigated the effects of intravitreally delivered AAV carrying a hybrid Flt-1 domain/human immunoglobulin G1 heavy chain Fc fragment. This approach has proved successful in various mouse models, as well as in an NHP model of ocular neovascularization,39,40 and the results were used to support initiation of phase 1/2 clinical trials (NCT01024998). A schematic of the different injection approaches is shown in Figure 1.
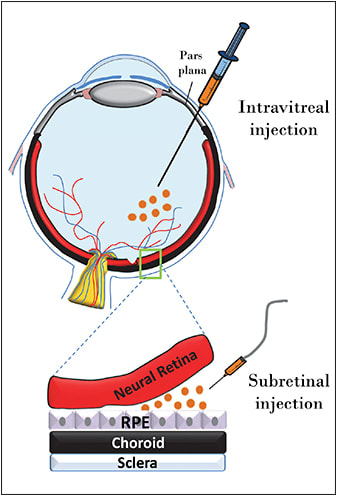
Figure 1. Schematic of subretinal vs intravitreal injection routes.
PHOTORECEPTOR-TARGETED THERAPIES ON THE HORIZON
Of the disease targets discussed above, none are associated directly with a genetic defect in the photoreceptors. Many animal models of photoreceptor-mediated disease have been corrected via AAV gene therapy in the laboratory, and several of these proof-of-concept studies are slated for clinical application in the next one to two years. Leading the charge are the following.
GUCY2D Leber Congenital Amaurosis-1
Mutations in GUCY2D (OMIM #600179) are associated with Leber congenital amaurosis-1 (LCA1), a severe early-onset disease characterized generally by profound cone dysfunction, abnormal rod function, nystagmus, loss of color vision, severely reduced VA, and normal retinal lamination, barring abnormalities in foveal cone outer segments and, in some patients, foveal cone loss.41
GUCY2D encodes retGC1, a protein that plays a pivotal role in the recovery phase of phototransduction. Subretinal delivery of AAV carrying Gucy2e (mouse homolog) led to long-term restoration of retinal function and useful vision and the preservation of retinal structure in multiple models of retGC1 deficiency over the long term.42-45
Because the disease target is the foveal cones, and unlike primates, mice lack a fovea (Figure 2), work is under way to characterize therapy in an all-cone model of retGC1 deficiency. Genzyme recently partnered with investigators at the University of Florida/University of Pennsylvania to conduct the final preclinical and safety studies necessary to file an IND and to initiate phase 1/2 clinical trials to evaluate the safety and efficacy of an AAV-based strategy for GUCY2D-LCA1.
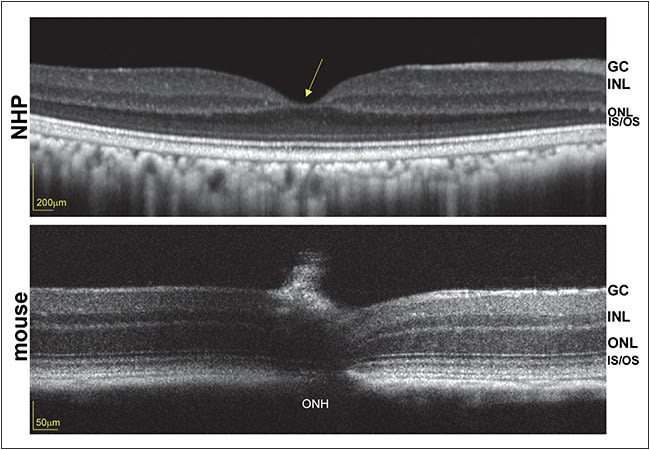
Figure 2. Optical coherence tomography scans of nonhuman primate (top) and mouse (bottom) retinas, obtained with Heidelberg Spectralis (Carlsbad, CA) and Bioptigen (Durham, NC), respectively. Foveal pit in the primate (yellow arrow) is absent from the mouse retina. ONH = optic nerve head, ONL = outer nuclear layer, INL = inner nuclear layer, GC = ganglion cell, IS/OS = inner segment/outer segment.
Achromatopsia
Mutations in cone-specific cyclic nucleotide gated channel α (CNGA3; OMIM #600053) and β (CNGB3; OMIM #605080), cone transducin α subunit (GNAT2; OMIM #139340), and the α subunit of cone-specific phosphodiesterase (PDE6C; OMIM #600827) are associated with the complete form of achromatopsia (ACHM), characterized by poor central VA, color blindness, and cone dysfunction.
Patients with ACHM can maintain a normal macular appearance, while some exhibit loss of central cones. AAV-mediated gene replacement has proved effective for improving retinal function and for useful vision in mouse models of GNAT2, CNGB3, and CNGA3 deficiency and in a dog model of CNGB3 deficiency.46-50
Work is under way to address therapy in a sheep model of CNGA3 deficiency (personal communication, 2014). The National Eye Institute, along with Applied Genetics Technologies Corporation, Inc. (AGTC; Alachua, FL), are currently funding natural history studies to evaluate patients with the CNGB3 form of the disease, as well as safety studies of an AAV-CNGB3 vector in anticipation of a phase 1/2 clinical trial. Efforts are also under way to address the CNGA3 form.
Recently, a detailed characterization of CNGA3 patients was published in which the investigators established testable efficacy endpoints in the anticipation of future AAV-mediated gene therapy trials.51
RPGR X-linked Retinitis Pigmentosa
Mutations in retinitis pigmentosa GTPase regulator (RPGR; OMIM #600827) are associated with ~70% of X-linked RP cases. RPGR encodes a protein by the same name, expressed in the photoreceptor cilia, which exhibits a mutational hotspot called ORF15.
Two naturally occurring canine models carrying mutations in ORF15 have been used to evaluate the effects of gene therapy. Mouse models of RPGR XLRP are also available, but they less efficiently mimic the human condition.52 Canine models (XLRPA1 and XLRPA2) exhibit slow or early-onset/rapid disease progression, respectively, thus providing insight into how patients with different causative mutations may respond.53
Investigators at the University of Pennsylvania/University of Florida have shown that subretinally delivered AAV-RPGR preserved photoreceptor structure and function over the long term.
Importantly, photoreceptor degeneration was completely arrested in affected dogs, contrasting the results seen in patients treated with AAV-RPE65, a therapy targeted to RPE rather than photoreceptors. AGTC has committed to a program of preclinical testing as a first step toward clinical trials to treat RPGR XLRP.
WHAT’S NEXT?
Intravitreal Delivery
Some LCA2 patients who received subretinal AAV-RPE65 underneath the fovea exhibited central retinal thinning and loss of VA.13 These outcomes raised the question of how to deliver genes to cells safely in fragile, diseased retinas that are prone to further damage upon surgically induced retinal detachment.
The ability to target genes safely to the foveal cone photoreceptors is especially significant because this area of the retina is responsible for acute, daylight vision. A recent push has been made to develop next-generation vectors capable of transducing photoreceptors from the vitreous, in the hopes that they could be administered, like the wet AMD drugs ranibizumab (Lucentis, Genentech, South San Francisco, CA) and bevacizumab (Avastin, Genentech), in an outpatient setting (Figure 1, page 21). While an intravitreal approach has been pursued to address wet AMD already, investigators relied on production of a secretable therapeutic protein (sFlt-1) by any transduced cell (often within the anterior segment). This sharply contrasts the ability to target transgene expression to distal retinal neurons.
Both rational design and directed evolution techniques have been used to design novel AAV capsid variants that exhibit increased photoreceptor transduction from the mouse vitreous.54,55 Their translation to NHP has proved challenging, however, due in part to anatomical differences between mouse and NHP eyes (Figure 2, page 22), dilution effects (higher vitreous volume in primates), and immunological differences across species. Further modifications to both AAV capsid and genome are being explored to overcome these hurdles.
Dual Vector
One drawback of AAV is its relatively small carrying capacity (~5,000 DNA bases). Developing gene therapies for diseases associated with mutations in large genes using AAV has therefore been challenging. To overcome this hurdle, dual AAV vector platforms have been developed, wherein large genes are split in half, and each half is delivered via a separate AAV vector.
Following coinfection, elements within each respective vector-delivered genome mediate recombination of the front and back half-sequences, leading to expression of functional, full-length protein. This strategy has proved useful for delivering full-length MYO7A (OMIM #276903) and ABCA4 (OMIM #601691) and for conferring therapy in models of Usher syndrome 1B and Stargardt disease, respectively.56-60
Importantly, it was shown that the sequence of recombined MYO7A transcript mediated by front and back half-vectors in vitro was 100% accurate, a result that will directly impact regulatory approval of such dual AAV vector platforms.57 Efforts are currently under way to increase the efficiency of dual AAV vectors and to characterize their safety profiles in vivo.
CONCLUSION
The list of recent successes in the field of retinal gene therapy is extensive. Clinical trials for RPE65-LCA2 paved the way for a multitude of AAV-based retinal gene therapy trials that have either already begun or are on the horizon. They also highlighted points to be considered as the field moves forward, such as the safety of submacular vector delivery, the differences between proof-of-concept findings in animal models vs patient outcomes, and therapeutic persistence.
There is still much work to be done, and if conducted cautiously, the future of AAV retinal gene therapy is sure to be bright. RP
REFERENCES
1. Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21:509-519.
2. Daya S, Berns KI. Gene therapy using adeno-associated virus vectors. Clin Microbiol Rev. 2008;21:583-593.
3. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92-95.
4. Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112-15117.
5. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240-2248.
6. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231-2239.
7. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979-990.
8. Banin E, Bandah-Rozenfeld D, Obolensky A, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther. 2010;21:1749-1757.
9. Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20:999-1004.
10. Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597-1605.
11. Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18:643-650.
12. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9-24.
13. Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital amaurosis type 2. Ophthalmology. 2013;120:1283-1291.
14. Cideciyan AV. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res. 2010;29:398-427.
15. Jacobson SG, Acland GM, Aguirre GD, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006;13:1074-1084.
16. Jacobson SG, Boye SL, Aleman TS, et al. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in Leber congenital amaurosis. Hum Gene Ther. 2006;17:845-858.
17. Kaiser J. Gene therapy. Two teams report progress in reversing loss of sight. Science. 2008;320:606-607.
18. Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4:120ra15.
19. Tolmachova T, Anders R, Abrink M, et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest. 2006;116:386-394.
20. Tolmachova T, Wavre-Shapton ST, Barnard AR, MacLaren RE, Futter CE, Seabra MC. Retinal pigment epithelium defects accelerate photoreceptor degeneration in cell type-specific knockout mouse models of choroideremia. Invest Ophthalmol Vis Sci. 2010;51:4913-4920.
21. Anand V, Barral DC, Zeng Y, et al. Gene therapy for choroideremia: in vitro rescue mediated by recombinant adenovirus. Vision Res. 2003;43:919-926.
22. Tolmachova T, Tolmachov OE, Wavre-Shapton ST, Tracey-White D, Futter CE, Seabra MC. CHM/REP1 cDNA delivery by lentiviral vectors provides functional expression of the transgene in the RPE of choroideremia mice. J Gene Med. 2013;91:825-837.
23. MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383:1129-1137.
24. Black A, Vasireddy V, Chung DC, et al. Adeno-associated virus 8-mediated gene therapy for choroideremia: preclinical studies in in vitro and in vivo models. J Gene Med. 2014;16:122-130.
25. Cereso N, Pequignot MO, Robert L, et al. Proof of concept for AAV2/5-mediated gene therapy in iPSC-derived retinal pigment epithelium of a choroideremia patient. Mol Ther. 2014 Apr 2. [Epub ahead of print]
26. Gal A, Li Y, Thompson DA, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270-271.
27. Vollrath D, Feng W, Duncan JL, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci U S A. 2001;98:12584-12589.
28. Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR. AAV-mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8:188-195.
29. Tschernutter M, Schlichtenbrede FC, Howe S, et al. Long-term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther. 2005;12:694-701.
30. Deng WT, Dinculescu A, Li Q, et al. Tyrosine-mutant AAV8 delivery of human MERTK provides long-term retinal preservation in RCS rats. Invest Ophthalmol Vis Sci. 2012;53:1895-1904.
31. Conlon TJ, Deng WT, Erger K, et al. Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Hum Gene Ther Clin Dev. 2013;24:23-28.
32. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427-1430.
33. Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci. 2007;48:1-10.
34. Yu H, Ozdemir SS, Koilkonda RD, et al. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol Vis. 2012;18:1668-1683.
35. Ellouze S, Augustin S, Bouaita A, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83:373-387.
36. Guy J, Qi X, Koilkonda RD, et al. Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci. 2009;50:4205-4214.
37. Yu H, Koilkonda RD, Chou TH, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci U S A. 2012;109:E1238-E1247.
38. Lai CM, Shen WY, Brankov M, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12:659-668.
39. Lai CM, Estcourt MJ, Wikstrom M, et al. rAAV.sFlt-1 gene therapy achieves lasting reversal of retinal neovascularization in the absence of a strong immune response to the viral vector. Invest Ophthalmol Vis Sci. 2009;50:4279-4287.
40. Lukason M, DuFresne E, Rubin H, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19:260-265.
41. Jacobson SG, Cideciyan AV, Peshenko IV, et al. Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet. 2013;22:168-183.
42. Boye SE, Boye SL, Pang J, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One. 2010 2010;5:e11306.
43. Boye SL, Conlon T, Erger K, et al. Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest Ophthalmol Vis Sci. 2011;52:7098-7108.
44. Mihelec M, Pearson RA, Robbie SJ, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. 2011;22:1179-1190.
45. Boye SL, Peshenko IV, Huang WC, et al. AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum Gene Ther. 2013;24:189-202.
46. Alexander JJ, Umino Y, Everhart D, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13:685-687.
47. Pang JJ, Deng WT, Dai X, et al. AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS One. 2012;7:e35250.
48. Carvalho LS, Xu J, Pearson RA, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20:3161-3175.
49. Komaromy AM, Alexander JJ, Rowlan JS, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19:2581-2593.
50. Michalakis S, Muhlfriedel R, Tanimoto N, et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. 2010;18:2057-2063.
51. Zelinger L, Cideciyan AV, Kohl S, et al. Genetics and disease expression in the CNGA3 form of achromatopsia: steps on the path to gene therapy. Ophthalmology. 2015 Jan 20. [Epub ahead of print]
52. Huang WC, Wright AF, Roman AJ, et al. RPGR-associated retinal degeneration in human X-linked RP and a murine model. Invest Ophthalmol Vis Sci. 2012;53:5594-5608.
53. Zhang Q, Acland GM, Wu WX, et al. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Human Mol Genet. 2002;11:993-1003.
54. Dalkara D, Byrne LC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76.
55. Kay CN, Ryals RC, Aslanidi GV, et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One. 2013;8:e62097.
56. Lopes VS, Boye SE, Louie CM, et al. Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 2013;20:824-33.
57. Dyka FM, Boye SL, Chiodo V, Hauswirth W, Boye SE. Dual AAV vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum Gene Ther Methods. 2014;25:166-177.
58. Colella P, Sommella A, Marrocco E, et al. Myosin7a deficiency results in reduced retinal activity which is improved by gene therapy. PLoS One. 2013;8:e72027.
59. Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118:1955-1964.
60. Trapani I, Colella P, Sommella A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2013;6:194-211.