







The Treatment of Choroidal Melanoma: How, Why, When and by Whom?
An ocular oncologist offers his clinical opinions.
Bertil Damato, MD, PhD, FRCOphth
The treatment of choroidal melanoma is challenging and controversial. Although the primary aim is to prolong life, almost 50% of patients die of their disease, despite apparently successful eradication of the ocular tumor.1
It is not known whether ocular treatment influences survival and if so in whom.2 Randomized clinical trials have been inconclusive.3 The lack of high-level evidence has resulted in speculative articles, some even suggesting that ocular treatment accelerates metastatic death.4-6
Most patients suffer some degree of visual loss after ocular treatment, and many also sacrifice the eye.7 There is a wide choice of therapeutic modalities, with endless debates as to which is “best.” Only a few randomized trials have been conducted, making it necessary to base treatment on the evidence provided by case series.
Patients and their relatives need expert help to cope with the many threats they must face: metastatic death, visual handicap, disfigurement, and all the implications of these outcomes. Clearly, there is more to treating these patients than just treating their choroidal melanoma.
There has recently been enormous progress in the management of these patients, with the development of specialized and well-equipped ocular oncology centers having highly skilled, multidisciplinary teams, including not only ocular and general oncologists but also geneticists, pathologists, radiotherapists, psychologists, specialist nurses, ethicists, and others.
With such expertise, it is possible to take full advantage of all the advances in ocular surgery, radiotherapy, molecular pathology, prognostication, and the treatment of metastatic disease. Opportunities for basic scientific research and clinical trials have also improved, enhancing the prospects for accelerated progress in this field.
Given such developments, the large majority of patients with choroidal melanoma treated in specialist centers do enjoy a good quality of life, also reporting high levels of satisfaction with the care they receive.
| Bertil Damato, MD, PhD, FRCOphth, is clinical lead in the Ocular Oncology Service, of the Royal Liverpool University Hospital in the United Kingdom and is honorary professor in the Department of Molecular and Clinical Cancer Medicine at the University of Liverpool. He reports no financial interests in any products mentioned in this article. Dr. Damato can be reached via e-mail at Bertil@Damato.co.uk. |
In this article, I describe an approach to the management of patients with choroidal melanoma, presenting the uncertainties that the clinician and the patient may face.
SCOPE OF OCULAR TREATMENT OF CHOROIDAL MELANOMA
Metastatic disease develops in almost all patients whose melanoma shows a class 2 gene expression profile, chromosome 3 loss, and BAP1 loss.8-13 This occurs despite ocular treatment, which therefore seems ineffectual at preventing metastatic spread (probably because it has already occurred). In these patients, the main benefits of ocular treatment are conservation of the eye and vision, if possible, and the prevention of pain and cosmetic loss.
Metastatic disease is exceptionally rare in patients who at the time of ocular treatment have a disomy 3, class 1 melanoma. It is not known, however, how many of these low-grade melanomas would undergo transformation to high-grade malignancy and metastasize if left untreated for any length of time.
Various authors have suggested that lethal and nonlethal choroidal melanomas are distinct from their inception and that fatal tumors metastasize long before the ocular tumor is detected and treated.14,15 Some melanomas may undergo malignant transformation and metastasize late, as suggested by a patient whose tumor suddenly grew dramatically into a mushroom shape after years of indolent behavior. Examination of the enucleated eye revealed low-grade, disomy 3 melanoma at the tumor base and high-grade, monosomy 3 melanoma at the apex.16 This patient later died of metastatic disease, which may have been prevented if treatment had been administered without delay.
Early treatment not only improves the chances of conserving vision, but might perhaps prolong life in some patients. When it is not possible to distinguish a large nevus from a small melanoma, a growing number of ocular oncologists are now performing biopsies to eradicate any malignancy without delay while avoiding unnecessary treatment of nevi (Figure 1).
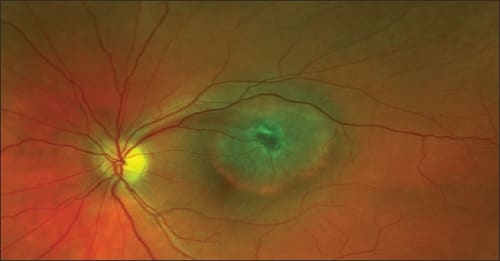
Figure 1. A 38-year-old woman was referred with a left, temporal choroidal tumor, measuring 5.9 x 5.1 x 1.4 mm, with a visual acuity of 20/20 but with a 6-month history of metamorphopsia. The patient was uncomfortable about the diagnostic uncertainty and unhappy about delaying treatment if the tumor was malignant. Trans-retinal biopsy confirmed that the tumor was a melanoma and she underwent proton beam radiotherapy. The visual acuity was 20/30 (92 letters) six months later. The patient accepts that the visual acuity is likely to deteriorate in time. Note the retinotomy from the biopsy, which was performed without any retinopexy or vitrectomy.
OCULAR TREATMENT Plaque Radiotherapy
In most centers, the first choice of treatment for choroidal melanomas is brachytherapy, usually delivered with an iodine-125 or ruthenium-106 plaque, other isotopes, such as palladium-103, being less common.17-19 If administered by an experienced surgeon with proper case selection, the results are good.
Accurate placement of the plaque can be difficult with small, posterior, and amelanotic tumors. To minimize the risk of tumor recurrence, it is conventional practice to position a suitably sized plaque so that it overlaps the entire tumor margin by at least 2 mm.20 With posterior tumors, this increases irradiation of the optic nerve and fovea, so that the temptation may be to reduce the radiation dose as much as possible, which increases the risk of under-treating the apical part of the tumor.
I resolve this dilemma by using beta-emitting ruthenium plaques, which have a limited range, and by developing simple tools and methods to position the plaque accurately, with its posterior edge at the posterior tumor margin so that a higher dose of radiation can safely be delivered (Figure 2).21-23 Others combine radiotherapy with phototherapy or replace the vitreous with silicone to attenuate the radiation.24,25
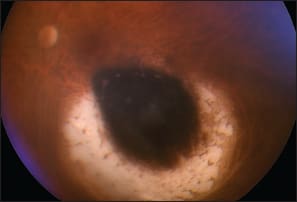
Figure 2. This 49-year-old woman was referred with a left, inferior, choroidal melanoma, which measured 10.7 x 9.6 x 3.4 mm. She was treated with an eccentrically positioned, 15 mm ruthenium-106 plaque, delivering 100 Gy to the tumor apex. After 6.4 years the tumor was inactive, with a thickness of 1.3 mm, and the visual acuity was 20/20-2 (98/100 letters). The author has learned that the tumoricidal effects of the brachytherapy extend beyond the visible choroidal atrophy.
Proton-beam Radiotherapy
Proton-beam radiotherapy is becoming more widely available as the number of cyclotron units increases. Proton-beam radiotherapy achieves high rates of local tumor control (ie >95%), even with tumors that are difficult to treat with a plaque.26,27 Some centers, therefore, use this modality for all patients, whereas others reserve it for tumors that cannot easily be treated by brachytherapy.
A total dose of 50-70 Gy is administered, usually in four fractions delivered on consecutive days. The treatment is planned using fundus photographs, ultrasound scans, ocular biometry, and radiographs of tantalum markers sutured to the sclera at known distances from the tumor and the limbus.
Protons release their destructive energy when they stop moving, this phenomenon being described as the “Bragg peak.” This peak is tall and narrow so that, in theory, the intervening healthy tissues are not damaged. In practice, however, this surface-sparing effect is usually reduced because it is necessary to “stretch” the peak into a plateau (by a spinning propeller within the beam, which slows down some protons more than others).
Collateral damage to the canaliculi, conjunctiva, and lacrimal gland can occur. Irradiation of the superior lid margin causes keratinization and pain on blinking. Associates and I have found that this problem can be avoided by treating through a closed eyelid.
Stereotactic Radiotherapy
With stereotactic radiotherapy, multiple finely collimated beams of radiation are “focused” from different directions onto the tumor, either concurrently or successively, so that the tumor receives much higher doses of radiation than the surrounding tissues. I have no personal experience with this technique, which is reported to produce similar results to proton-beam radiotherapy.28
Radiation-induced Complications
After radiotherapy, many patients experience visual loss, with some also developing a painful eye requiring enucleation.29-31 It is important to understand the pathophysiology of these complications and to distinguish between collateral damage and what I have termed the “toxic tumor syndrome.”
Collateral damage occurs when structures, such as the optic nerve, fovea, and lens, receive high doses of radiation. In contrast, toxic tumor syndrome arises as a result of necrosis and intratumoral radiation vasculopathy, which causes ischemia, vascular incompetence, and exudation which lead to macular edema, retinal exudates, retinal detachment, iris neovascularization, and painful neovascular glaucoma (Figure 3).32
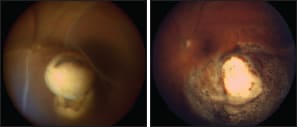
Figure 3. A 73-year-old man underwent proton beam radiotherapy for a left, inferior, choroidal melanoma, which measured 12.4 x 10.7 x 8.3 mm in size. The eye developed severe, exudative retinal detachment (A), which did not resolve with intra-vitreal anti-angiogenic agents but which was successfully treated by endoresection of the “toxic tumor” (B). Genetic studies showed no chromosome 3 loss, therefore indicating a good survival probability.
If the irradiated tumor is small, it may be possible to treat the patient successfully by applying transpupillary thermotherapy (TTT) or photodynamic therapy to the tumor or by intravitreal injections of steroids and/or an-tiangiogenic agents. Bulky toxic tumors may need to be removed by endoresection or exoresection, depending on their size and location.
Transpupillary Thermotherapy
With TTT, the tumor is heated to approximately 60°C for about one minute, so that its metabolic processes are disrupted irreversibly. This is done by directing 3-mm spots of infrared laser at the tumor using a slitlamp delivery system and contact lens. The intensity of the beam is adjusted so that the retinal vessels remain patent and retinal blanching does not occur for at least 45 seconds.
The treatment may need to be repeated several times until an atrophic choroidal scar is achieved. Initial enthusiasm has been tempered by relatively high recurrence rates so that this modality is usually administered as an adjunct to radiotherapy, except in patients with very small tumors (Figure 4).33
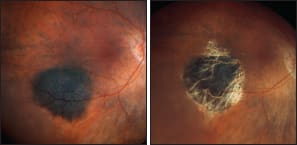
Figure 4. A 76-year-old man with diabetes mellitus was referred with a right, inferior choroidal tumor, which measured only 6.8 x 6.2 x 0.8 mm but which showed with orange pigment suggestive of malignancy (A). He was treated with transpupillary thermotherapy and 3.6 years later the tumor was inactive with visual acuity of 20/30 (92 letters) (B).
Photodynamic therapy
This treatment is administered using the same methods as for AMD. It is still an investigational technique as a primary procedure, with mixed results. PDT can reduce exudation from an irradiated tumor, improving vision.
Exoresection
Exoresection (eye wall resection, trans-scleral choroidectomy) involves the surgical resection of the tumor intact, through a large scleral trapdoor, without damaging the adjacent retina, and using hypotensive anesthesia to reduce hemorrhage.34 Adjunctive plaque radiotherapy is usually delivered after the excision to prevent local recurrence.
I perform this surgery for large tumors, when the chances of conserving vision are better than after radiotherapy (Figure 5).35, 36 As mentioned, exoresection is also useful for the toxic tumor syndrome. The most common complication is retinal damage, which occurs in about 15% of cases but which can be treated successfully by vitreoretinal surgery, thereby preventing retinal detachment. This operation is not widely performed as few ocular oncologists have opportunities to develop and maintain the required expertise.
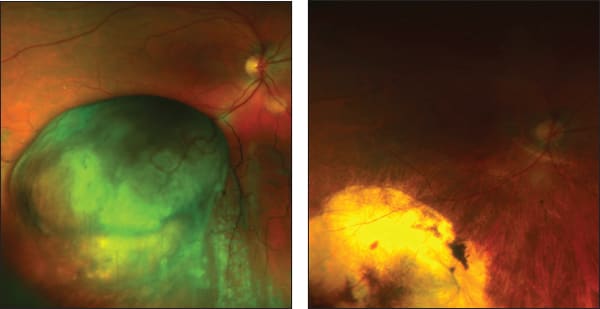
Figure 5. A 50-year-old man was referred with a right, inferior, collar-stud melanoma, measuring 10.2 x 9.5 x 12.0 mm and reducing the visual acuity to 20/50 (80 letters) (A). He was treated by exoresection and 1.7 years later the visual acuity was almost 20/20 (97 letters) (B).
Endoresection
With endoresection, the tumor is removed piecemeal with a vitreous cutter, which is passed through an incision in the overlying retina, unless a retinal flap is preferred. After tumor removal, endolaser is applied to ablate residual malignant cells and to achieve retinopexy.
To prevent postoperative hemorrhage and retinal detachment, the eye is then filled with silicone oil, which is removed after 12 weeks. Adjunctive radiotherapy can be administered as a secondary procedure if histology indicates high-grade malignancy. Local tumor control rates compare well with other forms of conservative therapy.37,38
Some authors, however, prefer to administer some form of neoadjuvant radiotherapy because of concerns about inducing tumor seeding and metastasis.39,40 Endoresection is also useful as a secondary treatment for recurrence or toxic tumor after radiotherapy (Figure 3).
Enucleation
Even where facilities for ocular conservation exist, about one-third of all patients require enucleation, usually because the tumor is large or involves the optic disc.7 Opportunities for ocular conservation may be lost because the tumor is not immediately detected when the patient presents with symptoms.41 Enucleation is performed in the standard manner, with the surgeon's preferred implant. Patients should be warned about phantom eye symptoms, which are common.42Biopsy
Biopsy
Increasingly, choroidal melanomas treated by radiotherapy or phototherapy are biopsied, to enhance prognostication by multivariate analysis integrating clinical, histologic, and genetic data.13 Genetic typing determines whether or not the tumor has metastatic potential and is performed using methods such as gene expression profiling, multiplex ligation-dependent probe amplification, and array comparative genomic hybridization.
If chromosome 3 loss and/or class 2 gene expression profile are absent, the survival prognosis is excellent even if the tumor is large (Figure 6). In patients with lethal genetic abnormalities, the likely survival time is estimated according to the clinical tumor stage, which indicates how long the tumor and any metastases have been growing and the histologic grade of malignancy, which correlates with tumor growth rate.43
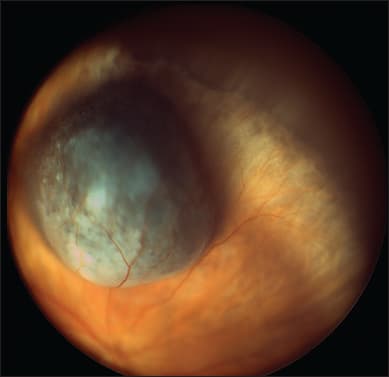
Figure 6. A 57-year-old man presented with a right, superior choroidal melanoma, which measured 15.2 x 13.9 x 5.2 mm. The tumor was treated with a 20 mm ruthenium-106 plaque. Trans-retinal biopsy with a vitreous cutter showed the tumor to be of mixed cell type with gains in the short arm of chromosome 6 and no chromosome 3 loss. Despite the large tumor size, the patient was given a good prognosis and 4.5 years later was alive with good local tumor control, no metastatic disease, and visual acuity of 20/30.
Associates and I have developed an online program for performing this multivariate analysis (available at www.ocularmelanomaonline.com). The sampling is done with a fine needle or vitreous cutter, transretinally or transsclerally ) (with or without a lamellar scleral flap), and at the time of the initial treatment or soon afterwards.44-46
With biopsy, most patients are found to have a “good melanoma” and are very relieved to know that their life expectancy is near normal. With regard to patients with poor prognoses, few regret their biopsy, because they feel empowered to prepare themselves and their families for any eventuality and to do whatever they can to improve their prospects for survival.47,48
Advances such as partial hepatectomy, microwave ablation, ipilimumab, isolated liver perfusion, and selective internal radiotherapy with yttrium beads have increased hopes for prolonging life in patients with metastasis. More studies are needed to evaluate such treatments.
Counseling
Patients with choroidal melanoma require skilled counseling throughout their care pathway. When first diagnosed, they need help to decide what form of management is most suited to their particular needs. I have for many years given patients an audio-recording of the discussion to help them remember what was said.49
As mentioned, patients and families can experience great distress, especially with pre-existing problems. Such patients can benefit from the care of a health psychologist.
CONCLUSION
The treatment of choroidal melanoma must be tailored to the size and location of the tumor and the needs and fears of the individual patient. Outcomes can be improved greatly by combining different therapeutic modalities. It is essential to ensure that the patient understands why the treatment is being administered, what can be achieved, and what is unattainable. I prefer to treat without delay in the hope of maximizing any opportunities for conserving vision and preventing metastasis.
Ocular treatment constitutes only one aspect of patient care. It is also essential to address any psychological problems. Of great concern is the threat of metastatic death. In most patients, biopsy and genetic tests will show this risk to be smaller than clinical features suggest, because lethal abnormalities are absent. When metastatic disease is anticipated, systemic screening may detect metastases early, possibly enhancing any opportunities to prolong life. Such care requires the involvement of a multidisciplinary team in a specialized ocular oncology center. RP
REFERENCES
1. Kujala E, Makitie T, Kivela T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003;44:4651-4659.
2. Damato B. Does ocular treatment of uveal melanoma influence survival? Br J Cancer. 2010;103:285-290.
3. Damato B. Legacy of the collaborative ocular melanoma study. A
4. Zimmerman LE, McLean IW, Foster WD. Does enucleation of the eye containing a malignant melanoma prevent or accelerate the dissemination of tumour cells. B
5. Manschot WA, Van Strik R. Is irradiation a justifiable treatment of choroidal melanoma? Analysis of published results. Br J Ophthalmol. 1987;71:348-352.
6. Damato B. Choroidal melanoma endoresection, dandelions and allegory-based medicine. Br J Ophthalmol. 2008;92:1013-1014.
7. Damato B, Lecuona K. Conservation of eyes with choroidal melanoma by a multimodality approach to treatment: an audit of 1632 patients. Ophthalmology. 2004;111:977-983.
8. Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12:461-468.
9. Scholes AG, Damato BE, Nunn J, Hiscott P, Grierson I, Field JK. Monosomy 3 in uveal melanoma: correlation with clinical and histologic predictors of survival. Invest Ophthalmol Vis Sci. 2003;44:1008-1011.
10. Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222-1225.
11. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410-1413.
12. Rummelt V, Folberg R, Woolson RF, Hwang T, Pe'er J. Relation between the microcirculation architecture and the aggressive behavior of ciliary body melanomas. Ophthalmology. 1995;102:844-851.
13. Damato B, Eleuteri A, Taktak AF, Coupland SE. Estimating prognosis for survival after treatment of choroidal melanoma. P
14. Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59:3032-3037.
15. Tschentscher F, Husing J, Holter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578-2584.
16. Callejo SA, Dopierala J, Coupland SE, Damato B. Sudden growth of a choroidal melanoma and multiplex ligation-dependent probe amplification findings suggesting late transformation to monosomy 3 type. A
17. Pe'er J. Ruthenium-106 brachytherapy. Dev Ophthalmol. 2012;49:27-40.
18. Finger PT, Chin KJ, Duvall G, Palladium-103 for Choroidal Melanoma Study Group. Palladium-103 ophthalmic plaque radiation therapy for choroidal melanoma: 400 treated patients. Ophthalmology. 2009;116:790-6,796.e1.
19. Jampol LM, Moy CS, Murray TG, et al. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS report no. 19. Ophthalmology. 2002;109:2197-2206.
20. Nag S, Quivey JM, Earle JD, et al. The American Brachytherapy Society recommendations for brachytherapy of uveal melanomas. Int J Radiat Oncol Biol Phys.2003;56:544-555.
21. Russo A, Laguardia M, Damato B. Eccentric ruthenium plaque radiotherapy of posterior choroidal melanoma. G
22. Damato B, Patel I, Campbell IR, Mayles HM, Errington RD. Visual acuity after Ruthenium(106) brachytherapy of choroidal melanomas. Int J Radiat Oncol Biol Phys.2005;63:392-400.
23. Damato B, Patel I, Campbell IR, Mayles HM, Errington RD. Local tumor control after 106Ru brachytherapy of choroidal melanoma. Int J Radiat Oncol Biol Phys. 2005;63:385-391.
24. Shields CL, Cater J, Shields JA, et al. Combined plaque radiotherapy and transpupillary thermotherapy for choroidal melanoma: tumor control and treatment complications in 270 consecutive patients. Arch Ophthalmol. 2002;120:933-940.
25. Oliver SC, Leu MY, DeMarco JJ, Chow PE, Lee SP, McCannel TA. Attenuation of iodine 125 radiation with vitreous substitutes in the treatment of uveal melanoma. Arch Ophthalmol. 2010;128:888-893.
26. Gragoudas ES. Proton beam irradiation of uveal melanomas: the first 30 years. The Weisenfeld Lecture. Invest Ophthalmol Vis Sci. 2006;47:4666-4673.
27. Damato B, Kacperek A, Chopra M, Campbell IR, Errington RD. Proton beam radiotherapy of choroidal melanoma: the Liverpool-Clatterbridge experience. Int J Radiat Oncol Biol Phys. 2005;62:1405-1411.
28. Dunavoelgyi R, Dieckmann K, Gleiss A, et al. Radiogenic side effects after hypofractionated stereotactic photon radiotherapy of choroidal melanoma in 212 patients treated between 1997 and 2007. Int J Radiat Oncol Biol Phys. 2012;83:121-128.
29. Desjardins L, Lumbroso-Le Rouic L, Levy-Gabriel C, et al. Treatment of uveal melanoma by accelerated proton beam. Dev Ophthalmol. 2012;49:41-57.
30. Fernandes BF, Weisbrod D, Yucel YH, et al. Neovascular glaucoma after stereotactic radiotherapy for juxtapapillary choroidal melanoma: histopathologic and dosimetric findings. Int J Radiat Oncol Biol Phys. 2011;80:377-384.
31. Wen JC, McCannel TA. Treatment of radiation retinopathy following plaque brachytherapy for choroidal melanoma. Cu
32. Damato B. Vasculopathy after treatment of choroidal melanoma. In: Joussen AM, Gardner TW, Kirchhof B, Ryan SJ, eds. Retinal Vacular Disease. Berlin, Germany; Springer; 2007:582-591.
33. Shields CL, Shields JA, Perez N, Singh AD, Cater J. Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology. 2002;109:225-234.
34. Damato BE. Local resection of uveal melanoma. Dev Ophthalmol. 2012;49:66-80.
35. Puusaari I, Damato B, Kivela T. Transscleral local resection versus iodine brachytherapy for uveal melanomas that are large because of tumour height. G
36. Bechrakis NE, Bornfeld N, Zoller I, Foerster MH. Iodine 125 plaque brachytherapy versus transscleral tumor resection in the treatment of large uveal melanomas. Ophthalmology. 2002;109:1855-1861.
37. Damato B, Groenewald C, McGalliard J, Wong D. Endoresection of choroidal melanoma. Br J Ophthalmol. 1998;82:213-218.
38. Garcia-Arumi J, Zapata MA, Balaguer O, Fonollosa A, Boixadera A, Martinez-Castillo V. Endoresection in high posterior choroidal melanomas: long-term outcome. Br J Ophthalmol. 2008;92:1040-1045.
39. Bechrakis NE, Foerster MH. Neoadjuvant proton beam radiotherapy combined with subsequent endoresection of choroidal melanomas. Int Ophthalmol Clin. 2006;46:95-107.
40. Schilling H, Bornfeld N, Talies S, et al. [Endoresection of large uveal melanomas after pretreatment by single-dose stereotactic convergence irradiation with the leksell gamma knife — first experience on 46 cases]. Klin Monbl Augenheilkd. 2006;223:513-520.
41. Damato EM, Damato BE. Detection and time to treatment of uveal melanoma in the United Kingdom: an evaluation of 2384 patients. Ophthalmology. 2012;119:1582-1589.
42. Rasmussen ML. The eye amputated — consequences of eye amputation with emphasis on clinical aspects, phantom eye syndrome and quality of life. Acta Ophthalmol. 2010;88 Thesis 2:1-26.
43. Eleuteri A, Damato B, Coupland SE, Taktak AFG. Enhancing survival prognostication in patients with choroidal melanoma by integrating pathologic, clinical and genetic predictors of metastasis. Int J Biomed Eng Technol. 2012;8:18-35.
44. McCannel TA, Chang MY, Burgess BL. Multi-year follow-up of fine-needle aspiration biopsy in choroidal melanoma. Ophthalmology. 2012;119:606-610.
45. Shields CL, Ganguly A, Bianciotto CG, Turaka K, Tavallali A, Shields JA. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology. 2011;118:396-401.
46. Sen J, Groenewald C, Hiscott PS, Smith PA, Damato BE. Transretinal choroidal tumor biopsy with a 25-gauge vitrector. Ophthalmology. 2006;113:1028-1031.
47. Cook SA, Damato B, Marshall E, Salmon P. Psychological aspects of cytogenetic testing of uveal melanoma: preliminary findings and directions for future research. Eye (Lond). 2009;23:581-585.
48. Beran TM, McCannel TA, Stanton AL, Straatsma BR, Burgess BL. Reactions to and desire for prognostic testing in choroidal melanoma patients. J Genet Couns. 2009;18:265-274.
49. Ah-Fat FG, Sharma MC, Damato BE. Taping outpatient consultations: a survey of attitudes and responses of adult patients with ocular malignancy. Eye (Lond). 1998;12:789-791.