







PEER REVIEWED
Understanding the Role of Bruch's Membrane in CNV
The more we know about retinal structure, the better our chances of treating disease.
Neda Barzegar-Befroei, MD • Tunde Peto, MD, PhD • Arthur A. Bergen, PhD • Imre Lengyel, PhD
Choroidal neovascularization1 is known to occur as an end result of nearly 40 ophthalmic diseases,2 including age-related macular degeneration.3 AMD is now reaching epidemic proportions, occurring in 0.7% to 1.4% of people aged 65 to 75 years old and in 11% to 16% of people over 85 years old.4 Approximately 10% of individuals with AMD develop (VEGF-driven) CNV.5,6 Prevention of CNV would be of great clinical significance in combating irreversible vision loss, but at best, anti-VEGF therapy halts the progression of the process only once it has already begun. There is currently no successful treatment to prevent its initiation.
There are several lines of recent evidence to support that Bruch's membrane (BM) plays a crucial role in CNV. It is believed that BM forms a physical barrier against the invasion of new vessels into the retina.7 With its strategic location between the retina and the choroidal circulation, BM is involved in the exchange of numerous biomolecules, oxygen, nutrients and waste products between the RPE and choriocapillaris. It plays a crucial role in cell-to-cell communication, cellular differentiation, proliferation, migration and tissue remodeling (see, for a recent review, Booij et al.8).
One of the initial steps in angiogenesis is the degradation of the discontinuous basal lamina of the choroidal endothelium,9 which forms part of BM. This step alone, however, is not sufficient for the development of CNV. The choriocapillaris can protrude into BM without leading to CNV in young and old patients, suggesting that neovascularization requires the degradation of the innermost aspects of BM.10 The biomechanical instability caused by the deposition of lipoprotein-derived debris, which contributes to the formation of basal linear deposits,11 is thought to facilitate new vessel growth, both horizontally (across BM) and vertically (within BM).12
Most likely, CNV is caused by a disturbed balance between a plethora of pro- and antiangiogenic factors.8 The picture is further complicated by the fact that normal aging, disease and/or environmental factors, such as diet, may (temporarily) influence the natural balance between pro- and antiangiogenic factors and that angiogenic factors may partly regulate each other's actions. A combination of these many factors presumably triggers a specific mechanism that leads to CNV.13
Here, we summarize the current understanding of the factors that may contribute to BM changes and progression to CNV.
GENETIC CONSIDERATIONS
There is ample evidence in the literature that patients with pathogenic DNA sequence changes (mutations or single nucleotide polymorphisms [SNPs]) may be more prone to CNV than their healthy counterparts. For example, patients with mutations in the metalloproteinase inhibitor 3 (TIMP-3) gene develop Sorsby's fundus dystrophy,14 a disorder resembling AMD, including drusen formation and submacular neovascularization.
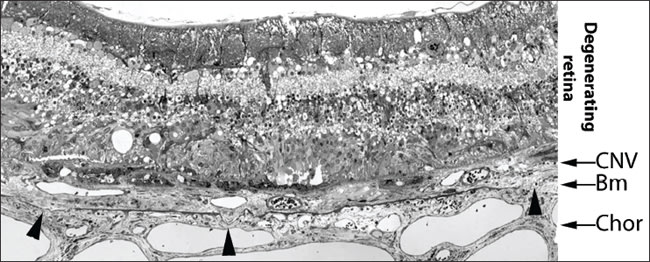
Figure 1. A light microscopic composite image of choroidal neovascularization (magnification: x600). Toluidine blue-stained semithin (0.7 µm) sections from an AMD patient show thinned, distorted or ruptured (labeled by arrow heads) Bruch's membrane (Bm) underlying exudates in the retina. Note the extensive degeneration of the neurosensory retina. Chor corresponds to choroid. Image courtesy of Professor Ulrich Schraermeyer, Center for Ophthalmology, University of Tübingen, Germany.
Relatives of AMD probands have a higher risk of developing AMD themselves.15 Indeed, common DNA sequence variants in or near the CFH, HTRA1-ARMS2, COL10A and VEGFA genes alter the risk for AMD.16 Patients with sporadic mutations in the CFH gene17 and fibulin-5 proteins are also more prone to developing AMD.18 Finally, the efficacy of anti-VEGF therapy in AMD may be partly depend on CFH, VEGF, and HTRA1 promoter genotypes.19 Thus, an inherited susceptibility component in the development of CNV should be considered.
STRUCTURAL CONSIDERATIONS
Wolfrum20 carried out a detailed light microscopic characterization of the bright line between the RPE and the choriocapillaris. This layer was originally termed the lamina vitrea because of its glassy appearance, but it was later renamed Bruch's membrane, honoring the German anatomist who first described it. This highly organized meshwork of connective tissues is composed of five layers: (1) the inner basal lamina of the RPE; (2) the inner collagenous zone (ICZ); (3) the elastic fiber layer; (4) the outer collagenous zone; and (5) the outer basal lamina of the choriocapillaris.21
Bruch's membrane thickness greatly varies among individuals22 and has been shown to double in size throughout life,23 from an average 2 µm in the first decade of life to 4.7 µm 80 years later.24 This thickening is caused by the deposition of waste products of the RPE, such as oxidized lipids and proteins. Over time, granular, membranous, filamentous and vesicular material accumulates into the different layers of BM.25
This accumulation eventually leads to the formation of focal or diffuse sub-RPE deposits in BM, best known as drusen.26 The deposition of biomolecules and the thickening of BM lead to functional changes in BM that have ramifications for the development of CNV as discussed below.
DEPOSITION OF PROTEOGLYCANS
Proteoglycans (PGs) are heavily glycosylated proteins that act as the “glue” of extracellular matrices, such as BM. PGs consist of a core protein covalently bonded to glycosaminoglycan (GAG) side chains. The functions of PGs differ depending on the type of core proteins and the type of GAG chains. PGs play important structural and filtration roles in BM through their interactions with other PGs, with collagen fibers and with hyaluronic acid.8
The negatively charged side chains cause PGs to bind water and positively charged cations, such as sodium, potassium and calcium, causing PGs to form a barrier for the passage of negatively charged macromolecules.27,28 Hence, PGs act as a barrier in BM.
RPE gene expression data suggest that the PG turnover rate is tightly controlled during life, with a constant ratio between the synthesis of heparan sulfate (25%) and chondroitin/dermatan sulfate (75%), the two main PGs incorporated into BM.29 However, after the age of 70 years old, there is a slight shift toward larger PGs, indicating a reduced ability of cells to digest the core molecules.27 Therefore, the heparan sulfate type of PG, with the slower turnover, becomes predominant in BM.8
This change in PG size and type will alter the function and permeability of BM, as described above. Additionally, it has been shown that heparan sulfate interacts with complement factor H, which is an important regulator of the complement cascade and local immune response.30 This interaction may be a key molecular switch that turns normal RPE aging into AMD pathology and subsequent CNV.8
DEPOSITION OF PROTEINS
With increasing age, the type 1, 3, 4 and 5 collagen content of BM increases,31,32 and the collagen fibers become coarse and irregular. This, together with increased cross-linking of the fibers, results in the decline in the solubility of the collagen.31,33 The amounts of non-collagen protein in BM also increase with age.31 The majority of these proteins accumulate as sub-RPE deposits and drusen, together with fats, carbohydrates34 and zinc.35
Proteomic studies have identified at least 129 different proteins,36 such as acute phase proteins, C-reactive proteins, complement components, complement inhibitors, apolipoproteins, beta amyloid, tissue inhibitor of metalloproteinase 3, vitronectin and complement C3, just to name a few.8 These proteins are derived from the RPE, blood and photoreceptors, supporting the hypothesis that both the RPE and the choroidal vasculature are involved in deposit formation.36
The presence of members of the complement cascade led to proposals that immune mediated–complement activation events are involved in AMD pathology and drusen formation in particular. This activation might be triggered by the accumulation of lipofuscin in the RPE37 and/or accumulation of oxidized lipids, beta amyloid protein or other pro-inflammatory “non-self ” molecules in BM.26,38,39 There is now mounting interest in genetic alterations in complement cascade molecules, such as CFH, C2/FB and C3, which, together, regulate the inborn basic activity level of the complement cascade in AMD.40-46
Figure 2. Protein deposition and alterations in vascular permability are among the earliest stages of the process that untimately leads to clinical signs of CNV in AMD. COURTESY OF PETER K. KAISER, MD
DEPOSITION OF LIPIDS
The lipid content of BM increases dramatically with age,47 eventually forming a lipid wall that impairs molecular transport across it.48 The majority of these lipids are esterified cholesterol, which shows sevenfold higher levels in the macula than in the periphery.49 Apart from its general accumulation in BM, esterified cholesterol is the most highly concentrated component of sub-RPE deposits.50
Cholesterol ester derivatives can generate oxidation by products that, together with other lipid-derived protein ad ducts, such as carboxyethylpyrrole (CEP), can crosslink proteins51, leading to the generation of insoluble, high molecular-weight aggregates found in sub-RPE deposits.52 These adducts not only crosslink proteins but can also trigger inflammatory reactions.53
The accumulation of lipids and the thickening of BM impair its ability to facilitate fluid and macromolecular exchange between the choroid and the RPE54 and delays oxygen delivery to the photoreceptors.12 In response to hypoxia, RPE cells overexpress VEGF,55 leading to CNV.56 Expression of VEGF is, among others, under the transcriptional regulation of hypoxia inducible factor-1α (HIF-1α), and this molecule is now emerging as an interesting therapeutic target for combating CNV.57
DEPOSITION OF AGEs
Advanced glycation end-products (AGEs) are chemically modified glycosylated or oxidized fats and proteins, produced either by external factors, such as smoking or cooking of food, or during the metabolism of fat, proteins and sugar.58,59 AGEs accumulate on cellular matrix proteins, such as collagens, in BM, which are resistant to proteolysis, to avoid catabolism. Here, they inhibit the proteins' functions and cause age-related damage.60
Additionally, high concentrations of AGEs in the serum or tissue activate the AGE receptor (RAGE), which has been associated with conditions such as atherosclerosis, diabetic nephropathy, and neurodegeneration through inflammatory and other mechanisms.61 In 2006, Yamada et al.62 showed that there is a more intense immune-staining for AGE receptors in areas containing basal deposits than in areas of normal BM, and therefore, these areas may play a role in development of AMD.
DEPOSITION OF METALS
• Calcification. Calcium has been shown to be deposited in BM.23 Studies have shown the extent of BM calcification to be positively correlated with age and later with AMD.63 A link between BM calcification and wet AMD could be explained by the notion that the calcification processes renders BM more brittle and more susceptible to breaks.
An interesting observation to support this idea is the findings of Bergen et al.64 and Le Saux et al.65 in 2000, in patients with pseudoxanthoma elasticum, an autosomalrecessive disease characterized by soft connective tissue calcification in, particularly, BM. In these patients, extensive calcifications of the elastic fibers of BM make the membrane brittle and prone to breaks. The breaks are visible on funduscopy as angioid streaks, which radiate from the macula toward the periphery66 and ultimately lead to neovascularization and loss of visual acuity.67
The mechanism responsible for calcium deposition in damaged or aging soft tissues involves a large number of environmental and genetic factors and is currently the focus of scientific investigations.8
• Iron and Copper. Iron is essential for many metabolic processes but can also cause damage by inducing local oxidative stress and generating reactive oxygen species.68 While a large network of molecules exists to maintain iron homeostasis, with age, iron accumulates in the body. The resulting overload of iron in the retina and RPE has been associated with AMD.69,71 Iron and its related compounds within the RPE can play a role in lysosome-mediated build-up of lipofuscin and, in general, cellular oxidative stress, aging, and apoptosis.72,73
On the surface of BM, iron and other metals are most likely involved in oligomerization of CFH molecules, affecting CFH's regulation of the complement system.74 The alternative pathway of the complement system has been implicated in AMD, as suggested by a large number of studies looking at polymorphisms in the CFH gene in particular.40-43
Of particular interest is the CFH Y402H allotype, which is a risk factor for AMD42 and which shows a higher propensity for self-association when exposed to copper.75 Patients carrying this polymorphism are also more likely to have classic CNV.76
• Zinc Deposition. Following observation of reduced zinc in the post mortem eyes of patients with AMD32 and a small zinc-supplementation trial,77 a beneficial effect for oral zinc supplementation in AMD was postulated. The results were confirmed by the AREDS trial.78 Subsequently, attention has been drawn to the fact that abnormally large quantities, in the range of millimoles, of zinc have been found in sub-RPE deposits and BM.35
A clear understanding of the role of zinc and its homeostasis is currently lacking. However, a study suggesting that zinc is involved in oligomerization of CFH74 may again implicate the alteration of the complement system in CNV.
LOSS OF BM INTEGRITY
Leukocytes, lymphocytes, macrophages, and endothelial cells may directly or indirectly degrade BM through the breakdown of collagens, thus facilitating or inducing CNV.79 Conversely, increased crosslinking can reduce flexibility and filtration capabilities, leading to decreased permeability of BM.31 This, in turn, may lead to BM breakage, facilitating endothelial cell migration and the development of CNV.
Interestingly, while the overall thickness of BM increases with age and disease, primarily due to the accumulation of fats, the elastic layer becomes thinner due to the decreasing elasticity of BM.7 In addition, breakdown of the elastic layer leads to this otherwise discontinuous layer being even less capable of limiting cell migration and the deposition of material to the outermost layers of BM.24
THE PROCESS
In summary, there is an age-related change in the molecular composition of BM. Besides alterations in PG content, a large number of RPE, choriocapillaris, and serum-derived proteins, fats and metal ions become trapped within BM, influencing its structure and function with undesirable repercussions, such as hypoxia, oxidative stress and inflammation.
The accumulation of biomolecules in BM causes changes in hydrostatic pressure and permeability,54 resulting in hypoxia and lack of nutrient exchange. This leads to the release of cytokines and growth factors, including VEGF, from the RPE cells. CNV is thought to occur due to imbalances between pro- and antiangiogenic factors.9 The age-related reduced integrity of BM and its breakdown by inflammatory cells further facilitate neovascularization.8
Oxidative stress, particularly in the macular region, where the photoreceptor density is highest, can be caused by the high metabolic rate of photoreceptors, the high local oxygen pressure, intense exposure to UV light, and the high metabolic rate of the RPE.44 With age-related deterioration in defense against oxidative stress, oxidative modifications of biomolecules can facilitate crosslinking, thus preventing the normal turnover that leads to drusen deposition.36
Drusen are found in the normal human eye with advancing age,80,81 but the presence of numerous hard and/or soft drusen (less well-defined borders, compared with hard drusen) in the macula is considered a risk factor for developing AMD.82 Drusen that take up fluorescein more readily and are presumed to be hydrophilic appear to predispose to CNV.83,84
Oxidative protein and lipid modifications in BM can trigger local immune response.26 The local complement response may initially be determined by the type of trigger, but the actual activity of the complement cascade is regulated by both genetic variations in regulatory proteins and their biochemical interactions.85
For example, complement factor H might interact with zinc in BM, leading to the deposition of large oligomers, probably into drusen precursors, loss of activity, and consequent deregulation of the complement cascade.74
While this may not be a complete list of what is happening in BM, the complexities of macro- and micromolecular interactions and the triggered physiological, or rather pathological, responses are complex and need to be elucidated further.
TREATMENT OPTIONS
At present, the full spectrum of BM changes that predispose the retina to neovascularization is unclear. As this review has shown, there are several factors that contribute to the aging of BM and, in ~10% of all AMD cases, to CNV.
Overproduction of VEGF is the most studied reason for development of CNV, and now, we have strategies to combat the disease at the early stages of neovascularization, using Lucentis, Avastin, photodynamic therapy or a combination of these. However, it would be ideal to interfere with processes that lead to CNV well before it starts. It appears we should slow the aging of BM or “rejuvenate” it to slow the progression toward CNV, geographic atrophy or both.
Successful preventive treatments rely on a proper understanding of the biochemical properties of BM, the interactions of BM with its microenvironment, and how these interactions change throughout life and disease course. While we have learned much about these mechanisms, there is plenty more to investigate. Nevertheless, the deposition of proteins, lipids, trace metals and other biomolecules that accumulate in BM appears to be an intriguing target for intervention at an early stage, before crosslinking by oxidation makes the process irreversible.
Limiting oxidative stress seems to be a primary target. Short-term supplementation with antioxidants has been tried, but has failed to slow the progression from asymptomatic drusen only to end-stage disease, though it successfully slowed the progression of CNV in a number of patients.78
Removal of accumulated trace metals is a new concept in combating BM changes.86 While it had not been tried for the eye, buffering (but not chelating) zinc in the brain in Alzheimer's disease can slow mental decline87 and may lead to better zinc distribution. Removal of iron (and perhaps copper) appears to help to limit oxidative damage, too.88 Removal of trace metals may also facilitate the dissolution of protein aggregates.74
How to facilitate appropriate handling of lipids has recently been reviewed by Curcio et al.,38 and promising targets are in the pipeline to inhibit AGE formation.89 It seems that the ultimate aim might be to try to resolve drusen. In fact, laser photocoagulation has been shown to resolve drusen successfully, unfortunately without reducing the risk of developing CNV or the loss of visual acuity.90
One could even argue that drusen create a protective environment, in which molecules can accumulate without directly evoking an inflammatory response, such as was shown for amyloid plaques in Alzheimer's disease.91 Thus, destroying drusen by laser could lead to excessive exposure to local inflammatory molecule, which could even worsen the problem.
It is clear that there is no such thing as a “magic bullet” in treating CNV or BM aging, but it is quite appropriate to say that a balanced diet and a healthy lifestyle (exercise and cessation of smoking) would help prevent, or at least delay, the onset of AMD and its most aggressive form, CNV. RP
REFERENCES
1. Green WR. The uveal tract: In: Spencer WH, ed. Ophthalmic Pathology: An Atlas and Textbook. 4th ed. Philadelphia, PA; W.B. Saunders; 1996:1597-1607.
2. Green WR, Wilson DJ. Choroidal neovascularization. Ophthalmology. 1986; 93:1169-1176.
3. Lopez PF, Grossniklaus HE, Lambert HM, et al. Pathologic features of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1991;112:647-656.
4. Chakravarthy U, Wong TY, Fletcher A, et al. Clinical risk factors for age-related macular degeneration: a systematic review and meta-analysis. BMC Ophthalmol. 2010;10:31.
5. Smith W, Assink J, Klein R, et al. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology. 2001;108:697-704.
6. la Cour M, Kiilgaard JF, Nissen MH. Age-related macular degeneration: epidemiology and optimal treatment. Drugs Aging. 2002;19:101-133.
7. Chong NH, Keonin J, Luthert PJ, et al. Decreased thickness and integrity of the macular elastic layer of Bruch's membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005; 166:241-251.
8. Booij JC, Baas DC, Beisekeeva J, Gorgels TG, Bergen AA. The dynamic nature of Bruch's membrane. Prog Retin Eye Res. 2010;29:1-18.
9. Folkman J. Angiogenesis: initiation and control. Ann N Y Acad Sci. 1982; 401: 212-227.
10. Guymer RH, Bird AC, Hageman GS. Cytoarchitecture of choroidal capillary endothelial cells. Invest Ophthalmol Vis Sci. 2004;45:1660-1666.
11. Curcio CA, Johnson M, Huang JD, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010;51:451-467.
12. Spaide RF, Armstrong D, Browne R. Continuing medical education review: choroidal neovascularization in age-related macular degeneration--what is the cause? Retina. 2003;23:595-614.
13. Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2011 Jul 23. [Epub ahead of print]
14. Weber BH, Vogt G, Pruett RC, Stöhr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby's fundus dystrophy. Nat Genet. 1994;8:352-356.
15. Assink JJ, Klaver CC, Houwing-Duistermaat JJ, et al. Heterogeneity of the genetic risk in age-related macular disease: a population-based familial risk study. Ophthalmology. 2005;112:482-487.
16. Yu Y, Bhangale TR, Fagerness J, et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum Mol Genet. 2011;20:3699-3709.
17. Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. 2011 Oct 23. [Epub ahead of print]
18. Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346-353.
19. McKibbin M, Ali M, Bansal S, et al. CFH, VEGF and HTRA1 promoter genotype may influence the response to intravitreal ranibizumab therapy for neovascular age-related macular degeneration. Br J Ophthalmol. 2011 May 10. [Epub ahead of print]
20. Wolfrum M. Zur Frage nach der Existenz des Glaskörperkanales. Graefes Arch Clin Exp Ophthalmol. 1908;67:370-376.
21. Sumita R. The fine structure of Bruch's membrane of the human choroid as revealed by electron microscopy. J Electronmicros. 1961;10:111-118.
22. Okubo A, Rosa RH Jr, Bunce CV, et al. The relationships of age changes in retinal pigment epithelium and Bruch's membrane. Invest Ophthalmol Vis Sci. 1999;40:443-449.
23. van der Schaft TL, Mooy CM, de Bruijn WC, Oron FG, Mulder PG, de Jong PT. Histologic features of the early stages of age-related macular degeneration. A statistical analysis. Ophthalmology. 1992;99:278-286.
24. Ramrattan RS, van der Schaft TL, Mooy CM, de Bruijn WC, Mulder PG, de Jong PT. Morphometric analysis of Bruch's membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci. 1994;35:2857-2864.
25. Huang JD, Presley JB, Chimento MF, Curcio CA, Johnson M. Age-related changes in human macular Bruch's membrane as seen by quick-freeze/deep-etch. Exp Eye Res. 2007;85:202-218.
26. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705-732.
27. Hewitt AT, Nakazawa K, Newsome DA. Analysis of newly synthesized Bruch's membrane proteoglycans. Invest Ophthalmol Vis Sci. 1989;30:478-486.
28. Rops AL, van der Vlag J, Lensen JF, et al. Heparan sulfate proteoglycans in glomerular inflammation. Kidney Int. 2004;65:768-785.
29. Booij JC, van Soest S, Swagemakers SM, et al. Functional annotation of the human retinal pigment epithelium transcriptome. BMC Genomics. 2009;10:164.
30. Meri S, Pangburn MK. Regulation of alternative pathway complement activation by glycosaminoglycans: specificity of the polyanion binding site on factor H. Biochem Biophys Res Commun. 1994;198:52-59.
31. Karwatowski, W.S, et al, Preparation of Bruch's membrane and analysis of the agerelated changes in the structural collagens. Br J Ophthalmol. 1995;79:944-952.
32. Newsome DA, Huh W, Green WR. Bruch's membrane age-related changes vary by region. Curr Eye Res. 1987;6:1211-1221.
33. Barnes MJ, Farndale RW. Collagens and atherosclerosis. Exp Gerontol. 1999;34:513-525.
34. Mullins RF, Hageman GS. Human ocular drusen possess novel core domains with a distinct carbohydrate composition. J Histochem Cytochem. 1999;47:1533-1540.
35. Lengyel I, Flinn JM, Peto T, et al. High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res. 2007;84:772-780.
36. Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682-14687.
37. Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182-16187.
38. Curcio CA, Johnson M, Rudolf M, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011 Sep 2. [Epub ahead of print]
39. Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:11830-11835.
40. Haines JL, Hauser MA, Schmidt S, et al, Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419-421.
41. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421-424.
42. Klein RJ, Zeiss C, Chew EY, Tsai JY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385-389.
43. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227-7232.
44. Scholl HP, Fleckenstein M, Charbel Issa P, Keilhauer C, Holz FG, Weber BH. An update on the genetics of age-related macular degeneration. Mol Vis. 2007; 13: 196-205.
45. Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of agerelated macular degeneration. N Engl J Med. 2007;357:553-561.
46. Despriet DD, van Duijn CM, Oostra BA, et al, Complement component C3 and risk of age-related macular degeneration. Ophthalmology. 2009;116:474-480.e2.
47. Pauleikhoff D, Harper CA, Marshall J, Bird AC. Aging changes in Bruch's membrane. A histochemical and morphologic study. Ophthalmology. 1990;97:171-178.
48. Hussain AA, Starita C, Hodgetts A, Marshall J. Macromolecular diffusion characteristics of ageing human Bruch's membrane: implications for age-related macular degeneration (AMD). Exp Eye Res. 2010;90:703-710.
49. Curcio CA, Millican CL, Bailey T, Kruth HS. Accumulation of cholesterol with age in human Bruch's membrane. Invest Ophthalmol Vis Sci. 2001;42:265-274.
50. Wang L, Clark ME, Crossman DK, et al, Abundant lipid and protein components of drusen. PloS One. 2010;5: e10329.
51. Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651-675.
52. Hollyfield JG. Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest Ophthalmol Vis Sci. 2010;51:1275-1281.
53. Hollyfield JG, Bonilha VL, Rayborn ME, et al, Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194-198.
54. Moore DJ, Hussain AA, Marshall J. Age-related variation in the hydraulic conductivity of Bruch's membrane. Invest Ophthalmol Vis Sci. 1995;36:1290-1297.
55. Schlingemann R. Role of growth factors and the wound healing response in agerelated macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2004;242:91-101.
56. Spilsbury K, Garrett KL, Shen WY, Constable IJ, et al. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am J Pathol. 2000;157:135-144.
57. Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K. Regulatory role of HIF-1a in the pathogenesis of age-related macular degeneration (AMD). Ageing Res Rev. 2009;8:349-358.
58. Baynes JW. The role of AGEs in aging: causation or correlation. Exp Gerontol. 2001;36:1527-1537.
59. Smit AJ, Lutgers HL. The clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr Med Chem. 2004;11:2767-2784.
60. Gugliucci A, Mehlhaff K, Kinugasa E, et al, Paraoxonase-1 concentrations in endstage renal disease patients increase after hemodialysis: correlation with low molecular AGE adduct clearance. Clin Chim Acta. 2007;377:213-220.
61. Sourris KC, Forbes JM. Interactions between advanced glycation end-products (AGE) and their receptors in the development and progression of diabetic nephropathy - are these receptors valid therapeutic targets. Curr Drug Targets. 2009;10:42-50.
62. Yamada Y, Ishibashi K, Ishibashi K, et al, The expression of advanced glycation endproduct receptors in rpe cells associated with basal deposits in human maculas. Exp Eye Res. 2006;82:840-848.
63. Spraul CW, Lang GE, Grossniklaus HE, Lang GK. Histologic and morphometric analysis of the choroid, Bruch's membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999;44(Suppl 1):S10-S32.
64. Bergen AA, Plomp AS, Schuurman EJ, et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228-231.
65. Le Saux O, Urban Z, Tschuch C, et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223-227.
66. Marmo MF, Wolfensbeger TJ. The Retinal Pigment Epithelium. New York, NY: Oxford University Press; 1998.
67. ZHu X, Plomp AS, van Soest S, Wijnholds J, de Jong PT, Bergen AA. Pseudoxanthoma elasticum: a clinical, histopathological, and molecular update. Surv Ophthalmol. 2003;48:424-438.
68. Rodriguez IR, Larrayoz IM. Cholesterol oxidation in the retina: implications of 7KCh formation in chronic inflammation and age-related macular degeneration.J Lipid Res.2010;51:2847-2862.
69. He X, Hahn P, Iacovelli J, et al. Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res. 2007;26:649-673.
70. Dunaief JL. Iron induced oxidative damage as a potential factor in age-related macular degeneration: the Cogan Lecture. Invest Ophthalmol Vis Sci. 2006; 47: 4660-4664.
71. Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch Ophthalmol. 2003;121:1099-1105.
72. Kurz T, Terman A, Brunk UT. Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys. 2007;462:220-230.
73. Kurz T, Karlsson M, Brunk UT, Nilsson SE, Frennesson C. ARPE-19 retinal pigment epithelial cells are highly resistant to oxidative stress and exercise strict control over their lysosomal redox-active iron. Autophagy. 2009;5:494-501.
74. Nan R, Gor J, Lengyel I, Perkins SJ. Uncontrolled zinc- and copper-induced oligomerisation of the human complement regulator factor H and its possible implications for function and disease. J Mol Biol. 2008;384:1341-1352.
75. Fernando AN, Furtado PB, Clark SJ, et al. Associative and structural properties of the region of complement factor H encompassing the Tyr402His disease-related polymorphism and its interactions with heparin. J Mol Biol. 2007;368:564-581.
76. Brantley MA Jr, Edelstein SL, King JM, Apte RS, Kymes SM, Shiels A. Clinical phenotypes associated with the complement factor H Y402H variant in age-related macular degeneration. Am J Ophthalmol. 2007;144:404-408.
77. Newsome DA, Swartz M, Leone NC, Elston RC, Miller E. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106:192-198.
78. Age-related Eye Disease Study Research Group, A randomised placebo controled, clinical trial of high-dose supplementation with vitamin Cand E, and beta carotene for age-related cateract and vision loss. Arch Ophthalmol. 2001;9:1439-1452.
79. Penfold PL, Provis JM, Billson FA. Age-related macular degeneration: ultrastructural studies of the relationship of leucocytes to angiogenesis. Graefes Arch Clin Exp Ophthalmol. 1987;225:70-76.
80. Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99:933-943.
81. Lengyel I, Tufail A, Hosaini HA, Luthert P, Bird AC, Jeffery G. Association of drusen deposition with choroidal intercapillary pillars in the aging human eye. Invest Ophthalmol Vis Sci. 2004;45:2886-2892.
82. De Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355:1474-1485.
83. Bressler SB, Maguire MG, Bressler NM, Fine SL. Relationship of drusen and abnormalities of the retinal pigment epithelium to the prognosis of neovascular macular degeneration. The Macular Photocoagulation Study Group. Arch Ophthalmol. 1990;108:1442-1447.
84. Pauleikhoff, D, et al, Drusen as risk factors in age-related macular disease. Am J Ophthalmol. 1990;109:38-43.
85. Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory statesthe examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age-related macular degeneration. Adv Immunol. 2007;96:141-177.
86. Lengyel I, Flinn JM, Peto T, et al. High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res. 2007;84:772-780.
87. Ritchie CW, Bush AI, Mackinnon A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685-1691.
88. Hadziahmetovic M, Song Y, Wolkow N, et al. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52:959-968.
89. Takeuchi M, Yamagishi S, Iwaki M, Nakamura K, Imaizumi T. Advanced glycation end product (age) inhibitors and their therapeutic implications in diseases. Int J Clin Pharmacol Res. 2004;24:95-101.
90. Parodi MG, Virgili G, Evans JR. Laser treatment of drusen to prevent progression to advanced age-related macular degeneration. Cochrane Database Syst Rev. 2009;3:CD006537.
91. Lee HG, Casadesus G, Zhu X, Takeda A, Perry G, Smith MA. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann N Y Acad Sci. 2004;1019:1-4.
| Neda Barzegar-Befroei, MD, and Imre Lengyel, PhD, are on the faculty of the University College London (UCL) Institute of Ophthalmology, where Dr. Lengyel is the Bill Brown Charitable Trust Senior Research Fellow. Tunde Peto, MD, PhD, serves both on the faculty of UCL and at the NIHR Biomedical Research Centre for Ophthalmology at Moorfields Eye Hospital NHS Foundation Trust, also in London. Arthur A. Bergen, PhD, is on the faculty of the Department of Molecular Ophthalmogenetics at the Netherlands Institute for Neuroscience and the Department of Clinical Genetics and Ophthalmology at the Academic Medical Center in Amsterdam. None of the authors reports any financial interest in any products mentioned in this article. Dr. Lengyel can be reached via e-mail at i.lengyel@ucl.ac.uk. This study was supported by the Bill Brown Charitable Trust (IL), the Henry Smith Charity (NB), the NIHR BMRC in Ophthalmology (TP), the Mercer Fund from Fight for Sight, and the Moorfields Eye Hospital Special Trustees (IL). |