







Treatment Approaches for Geographic Atrophy in AMD
A range of new therapeutic concepts show promise
Rajesh C. Rao, MD • Jennifer George • Netan Choudhry, MD
Age-related macular degeneration, the leading cause of severe vision loss in developed countries,1 encompasses an advanced non-exudative form known as geographic atrophy (GA), which accounts for 35% of all cases of late-stage AMD and 20% of legal blindness attribut able to AMD.2 In contrast to exudative (neovascular) AMD, GA manifests in older patients, and consequently, the prevalence is expected to rise as life expectancy increases.
The hallmark of GA is the depigmentation of the retinal pigment epithelium due to RPE dropout, resulting in sharply demarcated, round or oval regions of hypopigmentation within which choroidal vessels are more visible than adjacent regions (Figure 1); these atrophic areas are at least 175 μm in diameter.3 The atrophy is not limited to the RPE alone, but also involves the choroid and photoreceptors. These atrophic lesions exhibit loss of retinal sensitivity and correspond to absolute scotomata.4,5 The size of the lesions progresses over time to a rate of 1.5 to 2.1 mm2/year,6 usually beginning in the parafoveal retina with patches of GA coalescing, while new atrophic patches emerge.7 Once GA lesions involve the fovea, a striking loss of central vision results.
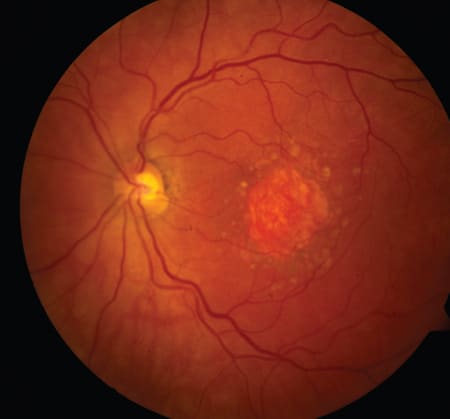
Figure 1. Geographic atrophy is characterized by round or oval regions of hypopigmentation within which choroidal vessels are more visible than adjacent regions.
GENETIC AND MOLECULAR PATHOPHYSIOLOGY OF GEOGRAPHIC ATROPHY
The molecular etiology of GA remains unclear. Genome-wide association studies (GWAS) analyzing variants in single nucleotide polymorphisms (SNPs) among large affected and nonaffected cohorts of patients with AMD have provided crucial insight into what the molecular underpinnings of AMD may be. Specifically, particular variants in genes encoding various components of the alternative complement pathway have been found to confer an increased risk of AMD, including complement factor H (CFH),8-11 complement component 2 (C2),12,13 complement factor B (BF),12,13 complement component 3 (C3),14,15 and complement factor I (CFI).16 Genetic association studies have revealed noncomplement, AMD risk-modifying variants as well, including ARMS2/HTRA1,17,18 fibulin 5,19 toll-like receptor-3 (TLR3),20 the hepatic lipase gene (LIPC),21 variant alleles near TIMP3,22 and loci associated with high-density lipoprotein metabolism.22
Of these genes, CFH, C3 and ARMS2/HTRA1 have been specifically implicated in GA susceptibility,23,24 as well as TLR3 in GA progression.20 GWAS have highlighted the importance of the alternative complement pathway, traditionally known to be an antibody-independent mechanism, whereby the innate immune system inhibits infection by specific pathogens. The mechanism by which this pathway increases the risk of developing GA continues to be investigated and may be related to the inability of mutant complement proteins to bind to modified heparin proteins present on Bruch's membrane and the choroid.25,26
Furthermore, components of the alternative complement pathway, including CFH and the membrane attack complex (C5b-9, responsible for the removal of pathogens and tissue damage by opsonization) have been localized to human RPE/choroid and drusen.11,27 ARMS2 is a protein expressed in the mitochondria of photoreceptors;28 therefore, mutant ARMS2-mediated mitochondrial activity may be related to the pathogenesis of GA. Injection of intravitreal ligands to induce TLR3 activation in wild-type mice resulted in RPE apoptosis and the development of lesions resembling GA. TLR3 knockout mice, however, were resistant to this effect.17
These results suggest that TLR3 activation is pathogenic to RPE and may be a therapeutic target for GA. Recently, decreased activity of DICER1, a microRNA-processing enzyme, has been linked to RPE degeneration accompanying GA in humans and laboratory mouse models.29 DICER1 activity prevented accumulation of the RPE-toxic Alu RNA,29 and overexpression of DICER1, via gene therapy, may be an approach to treating DICER1-deficient RPE in GA.
TREATMENT APPROACHES FOR GEOGRAPHIC ATROPHY
While the complete pathophysiological mechanism underlying GA has not yet been identified, key pathways that regulate the development of GA have been determined. Pharmacologic targeting of components of these pathways constitutes an important approach to developing novel therapies. Additionally, exciting advances in cell biology and gene and cytokine delivery have highlighted the promise of ocular gene therapy, growth factor–secreting implantable cell depots, and stem cell–derived RPE in treating GA.
Complement Factor Inhibitors
Complement factor inhibitors have been developed to modulate the activity of the alternative complement pathway. Ideally, the alternative complement pathway efficiently inhibits pathogens without damaging host tissue. In patients harboring particularly high-risk alleles, this balance may be tipped in favor of “collateral” damage to host cells and tissues, such as the RPE and choroid.30 The development of complement inhibitors to treat GA (as well as neovascular AMD) has emerged as an active area of drug development.
Phase 2 trials are enrolling patients to receive POT-4/Compstatin (Potentia Pharmaceuticals), a synthetic 13 amino acid cyclic peptide that binds C3, inhibiting activation of the complement cascade.30 TNX-234, a humanized monoclonal antibody fragment targeting complement factor D, a factor that promotes light-induced retinal damage,31 has been developed by Genentech and is enrolling GA patients for phase 1/2 trials.30
Already an FDA-approved drug for paroxysmal nocturnal hemoglobinuria, eclizumab (Alexion Pharmaceuticals) is a humanized anti-C5 antibody that has been approved by the FDA for clinical trials for GA.30
A pegylated aptamer–based C5 inhibitor called ARC1905 (Ophthotech) antagonizes the cleavage of C5 into C5a and C5b, thus inhibiting complement activation.30 Phase 1 results (presented at the Association for Research and Vision in Ophthalmology 2010 Annual Meet ing) showed that patients who received an intravitreal injection of ARC1905, when combined with ranibizumab for subfoveal neovascular AMD, experienced a 10-letter gain in visual acuity, and the drug was well tolerated.32
Several other complement pathway modifiers are in preclinical development, including TA106 (a Fab fragment of anti-CFB antibody), CR2-CFH hybrid proteins, JPE-1375 (small molecule C5aR peptidomimetic), anti-properdin (renders C3 converstase unstable), C1-INH (inhibits classical complement pathway), and sCR1 (a soluble form of endogenous complement receptor 1 that degrades active C3bBb).30
Finally, a recombinant form of CFH in preclinical development is intended to protect patients who harbor a pathogenic mutation in the CFH gene.30 The development of this new class of inhibitors represents a promising approach to reducing host tissue damage resulting from aberrant complement pathway activity.
Pharmacologic TLR3 Inhibition
TLR3 activation is associated with geographic atrophy.20 Interestingly, an agonist of TLR3 activation, double-stranded RNA (dsRNA), is typically an intermediate molecule encountered during viral infection. This report raises the possibility of a viral etiology for the development of GA and warns of the potential dangers of using smallinterfering RNA (siRNA, a type of dsRNA) in gene therapy approaches for eye disease.
Despite this warning, a recent study has implicated the kinases p38 and ERK1/2 in mediating the effects of TLR3 activation in triggering apoptosis of pigment cells.33 Thus, p38 and ERK1/2 inhibitors, such as SB-68132334 and vemurafenib,35 both of which have reasonable clinical safety pro files, may have a therapeutic effect in TLR3-mediated GA.
Visual Cycle Modulators
The regeneration of the visual pigment 11-cis-retinaldehyde (11-cis-RAL, the chromophore of rhodopsin) from all-trans-retinaldehyde (all-trans-RAL) constitutes an important path way in the visual cycle, and this mechanism represents a therapeutic, pharmacologic target for various forms of macular degeneration, such as GA. The conversion of 11-cis-RAL to all-trans-RAL is stimulated by the absorption of photons (light) within the outer segments of photoreceptors, and the regeneration of 11-cis-RAL from all-trans-RAL, which occurs mainly in the RPE, is essential for resensitizing photoreceptors to maintain continuous vision.48
In many forms of macular degeneration, such as Stargardt's disease and AMD, toxic lipofuscin pigments, such as N-retinylidene-N-retinyl-ethanolamine (A2E) and similar pro ducts, build up in the RPE.49 As light activation promotes the conversion of 11-cis-RAL to all-trans-RAL, A2E is formed as a byproduct. Agents that slow regeneration of 11-cis-RAL, such as isoretinoin (Roaccutane, Hoffman-La Roche), block formation of A2E in mouse models of Stargardt's disease, as well as age-dependent accumulation of lipofuscin in wildtype mice.
Indeed, isoretinoin-treated Stargardt's mice showed preser vation of light-stimulated activity in photoreceptors by electroretinography.50 Other agents, such as retinylamine, fenretinide, and small-molecule RPE65 antagonists, are potent visual cycle inhibitors of 11-cis-RAL regeneration that may find therapeutic application in GA and other forms of macular degeneration.51-53
Stem Cell–Derived RPE Transplantation
Retinal pigment epithelium degeneration is a hallmark of GA. Virtually unlimited numbers of RPE cells can be generated from autologous embryonic stem cell sources, in which donor skin is reprogrammed into induced pluripotent stem (iPS) cells and then coaxed into differentiating into functional RPE cells.36-38 Stem cell–derived RPE could then be transplanted subretinally as a suspension or sheet (after growth on a scaffold) to the areas of macular GA most at risk for progression. Transplantation of iPS cell–derived RPE into rat models of retinitis pigmentosa has been shown to maintain visual acuity.39
Limitations of this technique include the mixed success reported in clinical trials of the transplantation of RPE autologous sheets,40 in addition to the potential for nonterminally differentiated cells in the stem cell–derived RPE transplant to become tumorigenic. The success of stem cell–derived RPE transplantation will certainly be informed by the recently announced phase 1/2 trials, sponsored by Advanced Cell Technology and conducted at the Jules Stein Eye Institute at UCLA and Casey Eye Institute at Oregon Health & Science University, which will assess the effect of transplantation of ACT's proprietary human embryonic stem cell–derived RPE product in Stargardt's macular dystrophy.41
Growth Factor–Secreting Intraocular Implants
A recent report has detailed the results of a phase 2 trial of an encapsulated cell intraocular transplant for patients with GA.42 The intraocular transplant consists of encapsulated cells secreting ciliary neurotrophic factor (CNTF), a growth factor known to slow the loss of retinal neurons and photoreceptor outer segments during retinal degeneration.43,44 In this study, seven patients received sham surgery, seven received a low-dose CNTF-secreting intraocular implant (releasing 5 ng/day), and 10 received a high-dose CNTFsecreting intraocular implant (20 ng/day).
At 12 months, patients receiving the low- and high-dose CNTF implants experienced an increase in retinal thickness. Visual acuity stabilization (a loss of less than 15 letters) was noted in 96% of patients in the high-dose group, 83% in the low-dose group, and 75% in the sham group. Interestingly, a subgroup analysis of patients with baseline acuities of 20/63 or better revealed that 100% of patients in the high-dose group did not lose more than 15 letters, while only 57% patients in the combined low-dose and sham groups lost less than 15 letters. A mean 0.8-letter gain was seen in the high-dose group vs a mean 9.7-letter loss in the combined low-dose and sham groups.
However, there were no improvements or differences among the groups in Humphrey visual field test results or electroretinography sensitivity. The surgery and implantation were well tolerated over the one year of the study. While limited by relatively short follow-up and small patient group size, this trial represents an unusually promising step forward in the treatment of GA.
Gene Therapy
Gene therapy to a tissue of interest has seen early successes in the treatment of retinal disease. In particular, gene therapy for patients carrying a mutation in the RPE65 (Leber's congenital amaurosis) by subretinal injection of adeno-associated virus has been shown to be safe, with all patients having demonstrated improvement on subjective tests of visual acuity.45-47
DICER1, a microRNA processing enzyme, reduced activity in the retina of patients with GA, allowing buildup of toxic Alu RNA and resulting in RPE apoptosis. A vector-based approach to delivering more copies of the DICER1 gene and/or antisense oligonucleotides against Alu RNA to regions of GA may ameliorate the disease, although care should be taken to avoid inducing pathogenic dsRNA-mediated TLR3 activation.20
CONCLUSION
Currently, patients with GA do not have a targeted therapy capable of significantly improving visual function. With the explosion of genetic association studies and the advances in laboratory-based molecular biology, key genes, proteins and pathways that regulate susceptibility to and progression of GA are being uncovered at an exhilarating pace. A panoply of drugs that target the alternative complement pathway, TLR3 activation, and the visual cycle are in preclinical development, and early phase studies have shown that the drugs are safe and perhaps efficacious.
Innovations in cellular reprogramming techniques and stem cell biology have enabled the derivation of an unlimited number of autologous retinal cells from a simple skin biopsy, setting the stage for stem cell–derived RPE transplants for GA. Landmark successes in gene therapy for hereditary retinal degeneration have paved the way for the localized delivery of genes to rescue deficiencies in or aberrant activation of key GA proteins, such as DICER1. For the first time, a therapy, CNTF-secreting intraocular encapsulated cell technology, has improved vision in a subset of GA patients. The future is hopeful for new therapies that may prevent or retard GA and restore vision in those patients who suffer from geographic atrophy. RP
REFERENCES
1. van Leeuwen R, Klaver CC, Vingerling JR, et al. Epidemiology of age-related maculopathy: a review. Eur J Epidemiol. 2003;18:845-854.
2. Klein R, Klein BE, Knudtson MD, et al. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114:253-262.
3. Bird AC, Bressler NM, Bressler SB, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367-374.
4. Sunness JS, Gonzalez-Baron J, Applegate CA, et al. Enlargement of atrophy and visual acuity loss in the geographic atrophy form of age-related macular degeneration. Ophthalmology. 1999;106:1768-1779.
5. Schmitz-Valckenberg S, Bultmann S, Dreyhaupt J, et al. Fundus autofluorescence and fundus perimetry in the junctional zone of geographic atrophy in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:4470-4476.
6. Schatz H, McDonald HR. Atrophic macular degeneration. Rate of spread of geographic atrophy and visual loss. Ophthalmology. 1989;96:1541-1551.
7. Sunness JS, Bressler NM, Maguire MG. Scanning laser ophthalmoscopic analysis of the pattern of visual loss in age-related geographic atrophy of the macula. Am J Ophthalmol. 1995;119:143-151.
8. Edwards AO, Ritter R, 3rd, Abel KJ, et al. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421-424.
9. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419-421.
10. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in agerelated macular degeneration. Science. 2005;308:385-389.
11. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227-7232.
12. Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055-1059.
13. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458-462.
14. Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553-561.
15. Maller JB, Fagerness JA, Reynolds RC, et al. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200-1201.
16. Fagerness JA, Maller JB, Neale BM, et al. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100-104.
17. Yang Z, Camp NJ, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992-993.
18. Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet agerelated macular degeneration. Science. 2006;314:989-992.
19. Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346-353.
20. Yang Z, Stratton C, Francis PJ, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008;359:1456-1463.
21. Neale BM, Fagerness J, Reynolds R, et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U S A. 2010;107:7395-7400.
22. Chen W, Stambolian D, Edwards AO, et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A. 2010;107:7401-7406.
23. Scholl HP, Fleckenstein M, Fritsche LG, et al. CFH, C3 and ARMS2 are significant risk loci for susceptibility but not for disease progression of geographic atrophy due to AMD. PLoS One. 2009;4:e7418.
24. Magnusson KP, Duan S, Sigurdsson H, et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med. 2006;3:e5.
25. Herbert AP, Deakin JA, Schmidt CQ, et al. Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J Biol Chem. 2007;282:18960-18968.
26. Heurich M, Martinez-Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci U S A. 2011;108:8761-8766.
27. Anderson DH, Mullins RF, Hageman GS, et al. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411-431.
28. Fritsche LG, Loenhardt T, Janssen A, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892-896.
29. Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325-330.
30. Gehrs KM, Jackson JR, Brown EN, et al. Complement, age-related macular degeneration and a vision of the future. Arch Ophthalmol. 2010;128:349-358.
31. Rohrer B, Guo Y, Kunchithapautham K, et al. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci. 2007;48:5282-5289.
32. Cousins SW; Ophthotech Study Group. Targeting complement factor 5 in combination with vascular endothelial growth factor (VEGF) inhibition for neovascular age related macular degeneration (AMD): results of a phase 1 study. Paper presented at: Annual meeting of the Association for Research in Vision and Ophthalmology; Fort Lauderdale, FL; May 3, 2010.
33. Yu N, Zhang S, Sun T, et al. Double-stranded RNA induces melanocyte death via activation of Toll-like receptor 3. Exp Dermatol. 2011;20:134-139.
34. Sarov-Blat L, Morgan JM, Fernandez P, et al. Inhibition of p38 mitogenactivated protein kinase reduces inflammation after coronary vascular injury in humans. Arterioscler Thromb Vasc Biol. 2010;30:2256-2263.
35. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
36. Comyn O, Lee E, MacLaren RE. Induced pluripotent stem cell therapies for retinal disease. Curr Opin Neurol. 2010;23:4-9.
37. Kokkinaki M, Sahibzada N, Golestaneh N. Human induced pluripotent stemderived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native RPE. Stem Cells. 2011;29:825-835.
38. Osakada F, Ikeda H, Sasai Y, et al. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat Protoc. 2009;4:811-824.
39. Carr AJ, Vugler AA, Hikita ST, et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS One. 2009;4:e8152.
40. Falkner-Radler CI, Krebs I, Glittenberg C, et al. Human retinal pigment epithelium (RPE) transplantation: outcome after autologous RPE-choroid sheet and RPE cell-suspension in a randomised clinical study. Br J Ophthalmol. 2011;95:370-375.
41. Klimanskaya I, Hipp J, Rezai KA, et al. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using tran - scriptomics. Cloning Stem Cells. 2004;6:217-245.
42. Zhang K, Hopkins JJ, Heier JS, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108:6241-6245.
43. Li Y, Tao W, Luo L, et al. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PLoS One. 2010;5:e9495.
44. LaVail MM, Yasumura D, Matthes MT, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592-602.
45. Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest. 2011;121:2160-2168.
46. Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597-1605.
47. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240-2248.
48. Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cisvitamin A in the retinal visual cycle. Nat Genet. 1998;20:344-351.
49. Travis GH, Golczak M, Moise AR, et al. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469-512.
50. Radu RA, Mata NL, Nusinowitz S, et al. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci U S A. 2003;100:4742-4747.
51. Golczak M, Kuksa V, Maeda T, et al. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A. 2005;102:8162-8167.
52. Maiti P, Kong J, Kim SR, et al. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry. 2006;45:852-860.
53. Radu RA, Yuan Q, Hu J, et al. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49:3821-3829.
| Rajesh C. Rao, MD, is a vitreoretinal fellow at the Barnes Retina Institute of the Washington Univ. School of Medicine in St. Louis. Jennifer George is a freelance medical editor in Chicago. Netan Choudhry, MD, is a recent graduate of the Vitreoretinal Surgery Fellowship at the Massachusetts Eye & Ear Infirmary at Harvard Medical School in Boston. None of the authors reports any financial interest in any products mentioned in this article. Dr. Choudhry can be reached via email at netan.choudhry@gmail.com. |