







PEER REVIEWED
Pattern Dystrophy of the Retinal Pigment Epithelium
HISHAM ALKURAYA, MD ∙ KANG ZHANG, MD, PhD
Pattern dystrophy (PD) of the retinal pigment epithelium (RPE) refers to a heterogeneous group of dominantly inherited macular diseases characterized by the development of a variety of patterns of deposits of the yellow, orange, or gray pigment in the macular area (Figure 1).1,2 The better-known PDs include butterfly-shaped dystrophy, reticular dystrophy, multifocal pattern dystrophy simulating fundus flavimaculatus, adult vitelliform dystrophy, and fundus pulverulentus.1-7 The value of this clinical classification is questioned, however, since different types of PD are known to occur in different members of the same family carrying an identical mutation (see below). Furthermore, one form of PD can evolve into another within a single patient, and the type of PD may even be different between the two eyes of a patient.7,11-13 Therefore, PD should be considered as a single disease expressed in various manners.
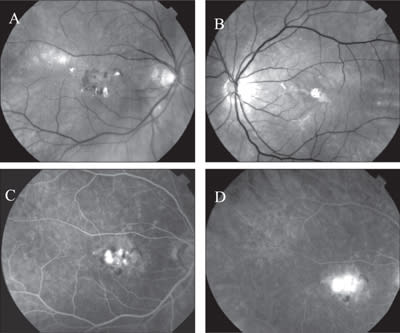
Figure 1. Red-free fundus photos of the right (A) and the left (B) eye in a patient with butterfly-shaped pattern dystrophy, complicated by a choroidal neovascular membrane in the right eye. Leaking CNV membrane is clearly demonstrated by FA (C and D).
Pattern dystrophies are known to be caused by various mutations in the human retinal degeneration slow (RDS)/peripherin gene on chromosome 6p21.2-cent, including a large deletion in exon3,13 missense mutations (Gly167Asp,14 Arg172Trp,15-18 Cys213Arg,18 Lys197Glu, Glu208Asp, Trp246Arg, Ser289Leu8, Cys213Tyr,19 and Cys250Phe20), nonsense mutations (Gln239ter and Tyr285ter),16 and a 2-base pair deletion affecting codons 299 and 300 with resulting frameshift (see Table 1).21 The RDS/peripherin gene encodes a photoreceptor-specific glycoprotein that may play a role in the development and maintenance of photoreceptor outer segment discs.22-24 Mutation in this gene could mediate PD disease pathogenesis by interfering with the integrity of the photoreceptor membrane.19
The age at onset in PD is highly variable, but patients tend to remain asymptomatic until the fifth decade, and some may remain asymptomatic. In general, mild impairment of central vision is first noticed in midlife.7 PD is classically described as having a “benign” course. However, several studies have shown that the disease can progress with age, and older individuals may exhibit atrophic depigmented lesions and/or choroidal neovascularization that can result in severe vision loss.9,12,19,20,25,26 (See Figure 1.)
Age-related macular degeneration (AMD) can be indistinguishable from the late appearances of PD since drusen often become less prominent or disappear in advanced stages of the disease. Furthermore, some patients with PD have deposits resembling drusen or, in fact, might have age-related drusen. Therefore, a small proportion of older individuals with PD may be misdiagnosed with AMD.20 Although usually confined to the macular area, peripheral retinal changes can also be observed in some patients. Occasionally, retinitis pigmentosa-like changes are seen in combi nation with PD;7 this illustrates that many “macular” dystrophies are indeed panretinal disorders at the molecular level.
BUTTERFLY-SHAPED PATTERN DYSTROPHY
Butterfly-shaped pattern dystrophy (BPD) was first described by Deutman et al.27 in a Caucasian family who displayed peculiar bilateral butterfly-shaped pigmentations on the macular region at the level of the RPE. Histopathological and electron microscopic examination of BPD-affected retina revealed an area of total loss of RPE and photoreceptor cell layer, with intact choriocapillaris and lipofuscin-containing cells in the subretinal space in the macula. Outside the area of RPE atrophy, the RPE is greatly distended by lipofuscin.19
In BPD, the central lesion is readily demonstrated by fluorescein angiography, which helps to distinguish this condition from other PDs of the macula.28 Fluorescein angiography usually shows early hyperfluorescence that outlines the hypofluorescent figures.22,28 On fundus autofluorescence, lesions in BPD may show increased as well as decreased autofluorescence, corresponding with changes in RPE lipofuscin within the lesion.29 Although Deutman et al.27 originally suggested a relatively benign course for the disease and described patients whose sole pathological features were abnormal electro-oculograms (EOGs), further studies described BPD as a chronic progressive disorder.
Patients are generally asymptomatic when diagnosed with BPD in their second or third decade and retain relatively normal visual acuity for most of their lives. However, the disease can progress with age, and older individuals may exhibit atrophic, depigmented lesions extending into the peripapillary region, with markedly reduced visual acuity.31 In general, patients exhibit normal dark adaptation, color vision, and full-field electroretinograms (ERG), have intact peripheral fields, and may have reduced EOG.13,14,27,28,30,31
RETICULAR DYSTROPHY OF THE RPE
Reticular dystrophy (RD) of the RPE was first described by Sjögren3 in 1950 as “dystrophia reticularis laminae pigmentosa retinae” — hence the name Sjögren reticular dystrophy. Clinically, RD's appearance is like a reticular network of darkly pigmented lines covering the posterior pole, with pigmented knots present at the intersection of the dark lines, resembling a fishing net with knots.3,7,32 The lesion usually starts at the fovea and then gradually extends to involve the whole posterior pole; it usually fades with age but may also be replaced by extensive atrophic changes in the RPE.7 Fluorescein angiography often shows the lesions better than ophthalmoscopy. FA shows clear hypofluorescent reticular net outlining areas of diffuse hyperfluorescence that correspond to zones of hypo- and hyperpigmentation, respectively.7,28,32 ERG in patients with RD is typically normal; however, both normal and abnormal EOG patterns have been reported.32
MULTIFOCAL PATTERN DYSTROPHY SIMULATING STARGARDT DISEASE
Multifocal pattern dystrophy simulating Stargardt disease/fundus flavimaculatus is characterized by irregular yellow-white flecks scattered throughout the posterior pole, often extending beyond the retinal vascular arcades7,33 (Figure 2). Macular abnormalities may range from various patterns of yellow or grayish deposits to well-demarcated lesions of severe chorioretinal atrophy.33 The flecks seen in multifocal pattern dystrophy resemble those encountered in fundus flavimaculatus, an autosomal recessive retinal dystrophy caused by mutation in the ABCA4 gene.34 On fluorescein angiography, these flecks are usually hyperfluorescent in the early and late phases. Contrary to most Stargardt cases, fluorescein angiography in multifocal pattern dystrophy generally does not show a so-called dark choroid.33
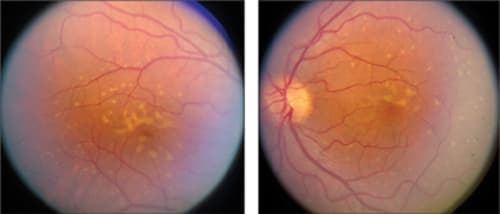
Figure 2. Color fundus photos showing retinal flecks in a patient with multifocal pattern dystrophy simulating Stargardt disease (STGD1)/fundus flavimaculatus.
On fundus autofluorescence imaging, the flecks show a highly increased autofluorescence, often with small adjacent zones of decreased autofluorescence.29 Unlike the other types of pattern dystrophies, this phenotype frequently displays signs of panretinal dysfunction, especially later in the course of the disease. A mild to severe constriction of the peripheral visual field is seen. Full-field ERG is often normal in early multifocal pattern dystrophy simulating Stargardt disease.35
With advanced disease, cone and rod function become compromised on the panretinal level, which is reflected by the appearance of full-field ERG abnormalities. Most patients show an abnormal EOG. On OCT, some of the Stargardt-like flecks appear as a highly reflective focal thickening of the hyper-reflective outer red line.36
Clinical findings that may help to distinguish this pattern dystrophy from Stargardt disease are the autosomal dominant pattern of inheritance, the relatively late onset (fifth decade), the comparatively good and stable visual acuity, and the absence of a “dark choroid.”29 However, the incomplete penetrance and the variable expression may mask the dominant pattern.
ADULT-ONSET FOVEOMACULAR VITELLIFORM DYSTROPHY
Adult-onset foveomacular vitelliform dystrophy (AFVD) is a polymorphic disease with clinical and genetic heterogeneity. Besides the peripherin/RDS gene, AFVD has been linked to mutations in VMD2 gene;37 it presents classically as bilateral, symmetrical, grayish-yellow, round, or oval-shaped lesions within the macular area.7,38 These lesions are mildly elevated and are typically one-third to one-half disc diameter in size but may occasionally be large and may thus be confused with Best vitelliform macular dystrophy.7,39,40
Fluorescein angiography typically shows hypofluorescence in the area corresponding to the vitelliform lesion, with a sur rounding ring of hyperfluorescence that increases in intensity in the late phases.7 In addition, indocyanine green angiography also shows either a central nonfluorescent spot or a diffuse hyperfluorescent area. Different fundus autofluorescence patterns have been reported, including normal, focal, patchy, ring-like, and linear, with inconsistent correlation with visual acuity41,42
Vitelliform lesions and their subretinal location can be clearly delineated using optical coherence tomography.41,43-46 Full-field ERG is typically normal, and contrary to Best vitelliform macular dystrophy, EOG is usually normal.47 Histopathological studies are inhomogeneous, depending on the stage of the disease at which the postmortem study was carried out,48-51 but they generally demonstrate large round pigment-laden cells in the subretinal space.
Vitelliform lesions are attributed also to extracellular material comprising photoreceptor debris, pigment granules, and RPE cells in various stages of disintegration.52 Unlike those in Best vitelliform macular dystrophy, the vitelliform lesions in patients with pattern dystrophy usually first appear in the fourth decade or beyond, are generally smaller, and do not show disruption in the layering of the yellow pigment in the dependent portion of the lesion.7
FUNDUS PULVERULENTUS
Fundus pulverulentus is probably the rarest type of all pattern dystrophies. It was first described by Slezak53 in 1969 and is characterized by coarse pigment mottling of the pigment epithelium in the macular area.7 Sometimes these pigmentary changes are impossible to differentiate from the ones seen in many other maculopathies, including AMD. Fluorescein angiography usually shows the hypofluorescent spots corresponding to the pigment mottling.25
SYSTEMIC ASSOCIATION
All main types of pattern dystrophy have been described in patients with Pseudoxanthoma elasticum, with fundus pulverulentus being the most common type.54 PDs have also been described in association with myotonic dystrophy, McArdle disease, maternally inherited diabetes, deafness, Crohn disease and others.55-58
CONCLUSION
In summary, PDs are a group of heterogeneous diseases that are primarily caused by mutations of RDS gene. Although they have historically been subdivided into different groups, keeping them under the umbrella of PD is clinically less confusing. PD is not necessarily visually benign, and progression can result in significant vision impairment and legal blindness in its advanced stages. Their phenotypic variation makes them sometimes difficult to diagnose, and they can easily be confused with other pigmentary maculopathies. Detailed history, including family history, combined with careful retinal exam, various methods of retinal imaging, and genetic testing, can lead to the proper diagnosis. RP
| Hisham Alkuraya, MD, practices in the vitreoretinal division of King Khalid Eye Specialist Hospital in Riyadh, Saudi Arabia, and is on the faculty in the Department of Ophthalmology at the University of California–San Diego. Kang Zhang, MD, PhD, is also on the faculty at the UCSD department of ophthalmology. Neither author reports any financial interest in any products mentioned in this article. Dr. Alkuraya can be reached via e-mail at halkuraya@gmail.com. |
REFERENCES
- Marmor MF, Byers B. Pattern Dystrophy Of The Retinal Pigment Epithelium. Am J Ophthalmol. 1976;13:112-116.
- Hsieh RC, Fine BS, Lyons JS. Pattern Dystrophy Of The Retinal Pigment Epithelium. Arch Ophthalmol. 1977;95:429-435.
- Sjogren H. Dystrophia Reticularis Laminae Pigmentosae Retinae. Acta Ophthalmol. 1950;28:279-295.
- Deutman A F, Rumke AML. Reticular Dystrophy Of The Retinal Pigment Epithelium. Arch Ophthalmol. 1969;82:4-9.
- Chopdar R. Reticular Dystrophy Of Retina. Br J Ophthalmol. 1976;60:342-344.
- Ayazi S, Fagan R. Pattern Dystrophy of Pigment Epithelium. Retina. 1981;1:287-289.
- Gass JMD. Stereoscopic Atlas Of Macular Disease. Philadelphia, Pa:Elsvier; 1997.
- Kingham JD, Fenzel RE, Willerson D, et al. Reticular Dystrophy Of The Retinal Pigment Epithelium. Arch Ophthalmol. 1978;96:1177-1184.
- Watzke RC, Folk JC, Lang RM. Pattern Dystrophy Of The Retinal Pigment Epithelium. Ophthalmology. 1982;89:1400-1406.
- Giuffre G, Lodato G. Vitelliform Dystrophy and Pattern Dystrophy Of The Retinal Pigment Epithelium: concomitant presence in a family. Br J Ophthalmol. 1986;70:526-532.
- Kim RY, Dollfus H, Keen TJ, et al. Autosomal Dominant Pattern Dystrophy of the Retina Associated with a 4-basepair insertion at Codon 140 in the Peripherin/RDS gene. Arch Ophthalmol. 113, 451-455.
- Yang Z, Li Y, Jiang L, Karan G, et al. A novel RDS/Peripherin gene mutation Associated with Diverse Macular Phenotypes. Ophthalmic Genet. 25:133-145.
- Fossarello M, Bertini C, Galantuomo MS, et al. Deletion in the Peripherin/RDS gene in Two Unrelated Sardinian Families with Autosomal Dominant Butterfly-Shaped Macular Dystrophy. Arch Ophthalmol. 1996;114:448-456.
- Nicholas BE, Sheffield VC, Vanderburgh K, et al. Butterfly-Shaped Pigment Dystrophy of the Fovea Caused by a point Mutation in codon 167 of the RDS gene. Nat Genet. 1993;3:202-207.
- Downes SM, Fitzke FW, Holder GE, et al. Clinical Features of Codon 172 RDS Macular Dystrophy: Similar Phenotype in 12 Families. Arch Ophthalmol. 1999;117:1373-1383.
- Kohl S, Christ-Adler M, Apfelstedt-Sylla E, et al. RDS/Peripherin Gene Mutations are Frequent Causes of Central Retinal Dystrophies. J Med Genet. 1997;34:620-626.
- Wells J, Wroblewski J, Keen J, et al. Mutations in the Human Retinal Degeneration Slow (RDS) gene Can Cause Either Retinitis Pigmentosa or Macular Dystrophy. Nat Genet. 1993;3:213-218.
- Payne AM, Downes SM, Bessant DA, et al. Founder Effect Seen in the British Population of the 172 Peripherin/RDS Mutation and Further Refinement of the Genetic Position of the Peripherin/RDS. Am J Hum Genet. 1998;62:192-195.
- Zhang K, Garibaldi DC, Li Y, et al. Butterfly-Shaped Pattern Dystrophy: A Genetic, Clinical, and Histopathololgical report. Ophthlmic Mol Genet. 2002;120:485-490.
- Frnacis PJ, Schultz DW, Gregory AM, et al. Genetic and Phenotypic Hetrogeneity in Pattern Dystrophy. Br J Ophthalmol. 2005;89:1115-1119.
- Nichols BE, Drack AV, Vandenburgh K, et al. A 2 Base Pair Deletion in the RDS gene associated with Butterfly-Shaped Pigment Dystrophy of the Fovea. Hum Mol Genet. 1993;2:601-603.
- Arikawa K, Molday LL, Molday RS, Williams DS. Localization of RDS/Peripherin in the Disk Membranes of Cone and Rod Photoreceptors: Relationship to Disk Membrane Morphogenesis and Retinal Degeneration. J Cell Biol. 1992;116:659-667.
- Connell G, Bascom R, Molday L, et al. Photoreceptor Peripherin is the Normal Product of the gene Responsible for Retinal Degeneration in the RDS Mouse. Proc Natl Acad Sci U S A. 1991;88:723-726.
- Travis GH, Sutcliffe JG, Bok D. The Retinal Degeneration Slow (RDS) Gene Product is a Photoreceptor Disc Membrane-associated Glycoprotein. Neuron. 1991;6:61-70.
- Parodi MB. Choroidal Neovascularizaiton in Fundus Pulverulentus. Acta Ophthlmol Scan. 2002;80:559-560.
- Feist RM, White MF Jr, Skalka H, Stone EM. Choroidal Neovascularazation in a patient with Adult Foveomacular Dystrophy and a Mutation in the Retinal Degeneration Slow Gene (Pro210Arg). Am J Ophthalmol. 1994;118:259-260.
- Deutman AF, Blommestein JD, Henkes HE, et al. Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthamol. 1970;83:558-569.
- Tuppuranen K, Mantyjarvi M. The importance of fluorescein angiography in diagnosing pattern dystrophies of the retinal pigment epithelium. Doc Ophthalmol. 1994;87:233-243.
- Boon CJ, den Hollnader AI, Hoyng CB. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008;213-235.
- Prensky JG, Bressnick GH. Butterfly-shaped macular dystrophy in four generations. Arch Ophthalmol. 1983;101:1198-1203.
- Pinckers A. Pattern dystrophy of the retinal pigment epithelium: a review. Opthalmic Paediatr Genet. 1988;9:77-114.
- Chen MS, Chang CC, Tsai TH, et al. Reticular dystrophy of the retinal pigment epithelium. J Formos Med Assoc. 2007;106:490-494.
- Boon CJ, van Schooneveld MJ, den Hollander AI, et al. Mutations in the peripherin/RDS gene are an important cause cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br J Ophthalmol. 2007;91:1504-1511.
- Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat.Gent. 1997;15:236-246.
- van Lith-Verhoeven JJC, van den Helm B, Deutman AF, et al. A peculiar autosomal dominant macular dystrophy caused by an asparagine deletion at codon 169 in the peripherine/RDS gene. Arch Ophthalmol. 2003;121:1452-1457.
- Ghazi NG, Dibernardo C, Ying HS, et al. Optical coherence tomography of enucleated human eye specimens with histological correlation:origin of the outer “red line”. Am J Ophthalmol. 2006;141:719-726.
- Krämer F, White K, Pauleikhoff D, Gehrig A, et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8:286-292.
- Renner AB, Tillack H, Kraus H, et al. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina. 2004;24:929-939.
- Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301-309.
- Boon CJ, Klevering BJ, den Hollander AI. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol. 2007;125:1100-1106.
- Furino C, Boscia F, Cardascia N, et al. Fundus autofluorescence, optical coherence tomography and visual acuity in adult-onset foveomacular dystrophy. Opthalmologica. 2008;222:240-244.
- Parodi MB, Iacono P, Pedio M, et al. Autofluorescence in adult-onset foveomacular vitelliform dystrophy. Retina. 2008;28:801-807.
- Pierro L, Tremolad G, Introini U, et al. Optical coherence tomography in adult-onset foveomacular vitelliform dystrophy. Retina. 2002;134:675-680.
- Benhamou N, Souied EH, Zolf R, et al. Adult-onset foveomacular vitelliform dystrophy: a study by optical coherence tomography. Am J Ophthalmol. 2003;135:362-367.
- Wirtitsch MG, Ergun E, Hermann B, et al. Ultrahigh resolution optical coherence tomography in macular dystrophy. Am J Ophthalmol. 2003;135:362-367.
- Querques G, Prato R, Iaculli C, et al. Correlation of visual function impairment and OCT findings in patients with Stargardt disease and fundus flavimaculatus. Eur J Ophthalmol. 2008;18:239-247.
- Theischen M, Schilling H, Steinhorst UH. EOG in adult vitelliform macular degeneration, butterfly-shaped pattern dystrophy and Best disease. Ophthalmologe. 1997;94:230:233.
- Gass JD. A clinicopathologic study of a peculiar foveomacular vitelliform dystrophy. Trans Am Ophthalmol Soc. 1974;72:139-156.
- Patrinely JR, Lewis RA, Font RL. Foveomacular vitelliform dystrophy, adult type. A clinicopathologic study including electron microscopic observation. Ophthalmology. 1985;92:1712-1718.
- Jaffe GJ, Schatz H. Histopathologic features of adult-onset foveomacular pigment epithelial dystrophy. Arch Ophthalmol 1988;106:958-960.
- Dubovy SR, Hairston RJ, Schatz H, et al. Adult vitelliform pigment epithelial dystrophy. Clinicopathologic correlation of three cases. Retina. 2000;20:638-649.
- Arnold JJ, Sarks J P, Killingsworth MC, et al. Adult vitelliform macular degeneration: a clinicopathological study. Eye. 2003;17:717-726.
- Slezak H, Hommer K. Fundus pulverulentus. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1969;178:176-182.
- Agarwal A, Patel P, Adkins T, Gass JD. Spectrum of pattern dystrophy in pseudoxanthoma elasticum. Arch Ophthalmol. 2005;123:923-928.
- Kimizuka Y, Kiyosawa M, Tamai M, Takase S.Retinal changes in myotonic dystrophy. Clinical and follow-up evaluation. Retina. 1993;13:129-135.
- Leonardy NJ, Harbin RL, Sternberg P Jr. Pattern dystrophy of the retinal pigment epithelium in patients with McArdle's disease. Am J Ophthalmol. 1988;106:741-742.
- Massin P, Virally-Mond M, Vialettes B, et al. Prevalence of macular pattern dystrophy in maternally inherited diabetes and deafness. GEDIAM Group. Ophthalmology. 1999;106:182-187.
- De Franceschi P, Costagliola C, Soreca E, et al. Pattern dystrophy of retinal pigment epithelium in Crohn's disease. A case report. Ophthalmologica. 2000;214:441-446