







Clinical Management of Systemic Inflammatory Diseases Affecting the Posterior Segment
Part 1 of this two-part series explores chorioretinal involvement in GCA, Adamantiades-Behçet's disease, SLE and APS.
THOMAS E. FLYNN, MD • GALE A. McCARTY, MD, FACP, FACR
A large number of immune-mediated diseases affect the retina and its vascular structures, along with other organs and systems in the body.1 The ophthalmologist examining the retina has a unique opportunity to view functioning vessels and nervous tissue and accurately evaluate their states, both in health and disease processes. The importance of accurately assessing the retina and diagnosing underlying vasculitic or vasculopathic disorders extends beyond treating a potentially blinding disease to making a diagnosis that can save a patient's life. Recognizing underlying patterns of retinal neuronal and vascular damage can facilitate proper laboratory evaluation, initiation of prompt consultations with other specialists, and inception of an effective systemic treatment regimen for the eyes and the rest of the body.
This article will focus on four such entities: giant cell arteritis, Adamantiades-Behçet's disease, systemic lupus erythematosus and a more recently characterized entity, antiphospholipid antibody syndrome. We review the diagnostic criteria, laboratory workup, ocular and retinal involvement and current therapy for each of these diseases; newer concepts in the immunopathogenesis of these diseases will also be covered briefly. The intent of this article is to provide a guide to rapid movement from the ophthalmoscope to the initiation of a treatment protocol.
GIANT CELL ARTERITIS
Giant cell arteritis (GCA), or temporal arteritis, is an inflammatory disorder involving large and medium-size arteries with muscle and internal elastic lamina in their walls, such as the temporal artery or the ophthalmic artery and its tributaries, including the posterior ciliary arteries and the retinal artery. Together, these arteries supply the optic nerve and most of the inner retina,2,3 and they are involved in arteritic anterior ischemic optic neuropathy.
Giant cell arteritis is estimated to occur primarily in whites of Northern European heritage, with an incidence of 25 cases per 200,000 persons aged 50 or older, which increases by each decade over 50. It classically presents as a unilateral headache with varying degrees of touch tenderness over the temporal artery just anterior to the ear and extending upward over its branches, with sudden onset of decreased visual acuity, described as a curtain descending over, or a graying of, the visual field. Associated features are jaw and scalp claudication and, less commonly, cognitive difficulties, weight loss, or arterial involvement internally. Muscular pain limited to the shoulder and hip girdle without visual symptoms or temporal artery tenderness, called polymyalgia rheumatica (PMR), may precede full blown GCA.4
Key laboratory studies include a high sensitivity C-reactive protein (CRP), an erythrocyte sedimentation rate (ESR), upper limit of normal is 100 mm, value affected by coincident anemia), or preferably a Westergren sedimentation rate (WSR), upper limit of normal is 200 mm, value not affected by anemia and a better test.5 These tests must be obtained prior to initiation of steroid therapy to be a useful baseline from which future therapeutic decisions can be made. As CRP reflects tissue necrosis, while ESR is more reflective of fibrinopeptides made in the liver, these represent somewhat different mechanisms of inflammation, do not always correlate, and have different rates of response to treatment. The most severe ocular complications tend to occur with ESRs between 70 and 100 mm in a large series of 273 patients. A complete blood count may show leukocytosis or be normal. A complete metabolic profile may show nonspecific elevation of transaminases.6
The classical ocular manifestation of GCA is anterior ischemic optic neuropathy (AION) with evidence of arteritis (arteritic AION), with elevated inflammatory parameters (ESR, WSR, CRP). The patient may manifest pre-existing systemic complaints such as PMR, jaw claudication, pain and tenderness to touch in one or both temples, and weight loss due to decreased appetite or jaw pain. The onset of vision loss is usually uniocular, painless and profound, with a very limited window for initiation of steroid treatment before permanent loss of vision occurs in one or both eyes.7-9
Atypical presentations of temporal arteritis with bilateral onset,10 no premonitory systemic symptoms, normal blood tests,11 and lesser visual symptoms have been described. Obtaining a timely temporal artery biopsy remains the standard diagnostic maneuver to prove this disease, but like the blood tests that are usually obtained, false negative or equivocal results can occur with these biopsies. Currently, neither immunophenotyping of WBC around the vessel nor endothelial cell immunomarkers offers additional diagnostic or prognostic benefit, but these are under development.
A high clinical index of suspicion, bolstered by the findings on ophthalmoscopy, frequently leads to initiation of systemic steroids, even in the absence of clear confirmatory laboratory or pathology results. The usual ophthalmoscopic finding in GCA is AION with a white optic nerve, swelling of all or part of the circumference of the disc, loss of the optic cup, and surrounding retinal edema appearing to be either a central or branch retinal artery occlusion (CRAO or BRAO).8 The involved retina, particularly around the optic nerve, appears white and swollen, with obvious attenuation of the arteriolar vessels leaving the nerve. A cherry-red spot may be seen, characteristic of a CRAO, or segmental loss of retinal anatomy consistent with a BRAO is noted. An example of GCA retinopathy can be seen in Figure 1 below.
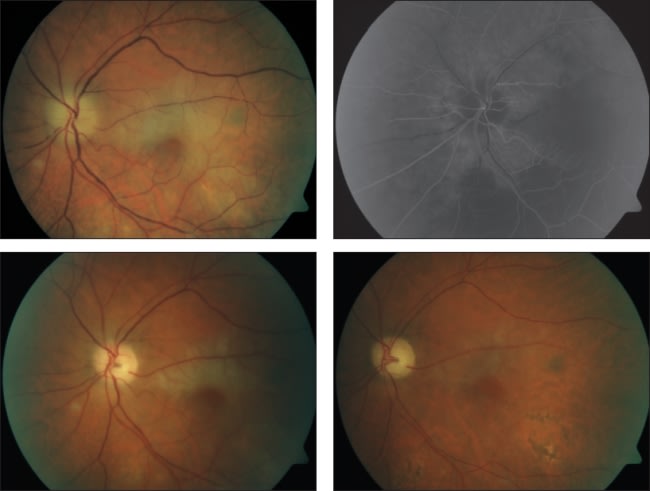
Figure 1. Images from an 80-year-old male with giant cell arteritis suffering from polymyalgia rheumatica. Fundus photo (top left) and fluorescein angiogram (top right) show poor circulation around the optic nerve. One week after administering highdose oral prednisolone, fundus photo (bottom left) shows improved condition. One month after initiation of steroid (bottom right), the condition continues to improve.
An atypical pattern may involve a more subtle abnormality of the optic nerve morphology with a simultaneous cilioretinal artery occlusion.12 Cotton wool spots are sometimes seen acutely in the evolution of arteritic AION;13,14 given the predilection of GCA for large and medium-size arteries, these lesions are most likely due to platelet aggregation causing embolization of capillaries in the retina.
More profound visual loss can result from a loss of choroidal circulation, indicating involvement of the more proximal ophthalmic artery rather than the retinal artery.15 In some cases, posterior ischemic optic neuropathy (PION) is the primary cause of profound visual loss, and the modest associated retinal findings can be difficult to evaluate.16 In many cases, a fluorescein angiogram may be indicated to assess the degree of damage to the optic nerve and retinal vasculature, even when the diagnosis has been made.
In rare cases, the first presentation of GCA and AION is as an advanced ocular ischemic syndrome in one or both eyes with hypotony, severe loss of retinal circulation, and neovascularization, uveitis and rubeosis iridis;17,18 by implication, the profound vision loss is not noted at onset and underlying systemic symptoms do not occur or are not successfully treated. Case reports of retinal vasculitis19 and a panuveitis simulating a multifocal choroiditis20 have also been described due to GCA.
Involvement of the choroid in GCA is common, ranging from slowing of the choroidal circulation time seen on indocyanine green (ICG) and fluorescein angiography21 to loss of choroidal circulation due to ophthalmic artery occlusion, with long-term bone spicule pigmentation. Linear choroidal scars, known as Siegrist streaks,22 were reported in a patient's right eye one month after loss of vision due to AION. Loss of vision can also occur in patients on longterm corticosteroid therapy due to drug-induced central serous chorioretinopathy.23
Making a timely diagnosis of GCA is critical as systemic steroid therapy, begun promptly and aggressively, can limit the degree of nerve fiber and vision loss in one eye and prevent a bilateral occurrence of the ischemic optic neuropathy. Unfortunately, the likelihood of recovery of vision once it is lost is limited,9 even in the setting of intravenous pulse methylprednisolone treatments. The importance of long-term, slowly tapered systemic steroids24 is emphasized by occasional reports in the literature and the authors' experience of AION recurring years after cessation of steroids for "successful" treatment of GCA and associated AION, with further vision loss in an already affected or contralateral eye. Concomitant aspirin therapy is also important.
Given the significant morbidity of long-term systemic steroids in an elderly and debilitated population, efforts have been made to study various steroid-sparing agents, such as methotrexate, dapsone, or even anti–tumor necrosis factor (TNF)-α agents to allow for more rapid tapering off of steroids. To date, the success of these efforts has been modest.
ADAMANTIADES-BEHÇET'S DISEASE
Adamantiades-Behçet's disease (ABD) is a progressive, devastating and often fatal multisystem disease25 with a significant predilection for affecting the retina and its circulation, in addition to the well-known hypopyon panuveitis that characterizes this disorder. ABD is diagnosed clinically, most often by the International Study Group criteria, in which recurrent apthous ulceration is the major criterion, with two of the following: recurrent genital ulceration, ocular lesions (anterior/posterior uveitis, vitritis, or retinal vasculitis — see Figure 2, below), skin lesions (erythema nodosum, pseudofolliculitis/papular/acneiform lesions), and pathergy (the formulation of a pustule at a venipuncture or needle-stick site 24 to 48 hours out).26 These findings are subject to exclusions and should be confirmed by a physician. While this disease is not diagnosed from laboratory data, a number of tests may be of value in bolstering an equivocal case or one with incomplete criteria for diagnosis.
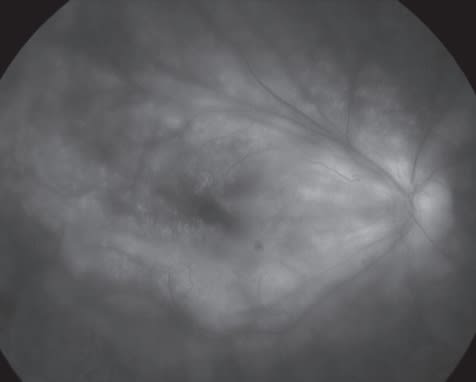
Figure 2. Late-phase fluorescein angiogram demonstrating retinal vasculitis in a patient with Adamantiades-Behçet's disease.
Adamantiades-Behçet's disease is one of the few retinal vasculitides characterized by retinal arteritis as much as sheathing of venules: The appearance of the retina may be confused with a BRAO or, in extensive involvement of arterioles, with a CRAO. A mixed arteriolar-venular occlusive disease27 may be seen, and involvement of the optic nerve may mimic AION28 and PION29 in some cases. Macular changes including macular star30 and serous detachment31 have been reported.
On fluorescein angiography, features of ABD include widespread leakage of dye from retinal vessels and the optic nerve and macula32 and profound and rapidly evolving destruction of the capillary net. Involvement of the retinal circulation can lead to disc33 and retinal neovascularization and vitreous hemorrhage,34 with their attendant long-term complications. A pattern of retinal swelling and hemorrhage can be seen that is sometimes confused with infectious (viral or luetic) retinitis, particularly when accompanied by severe panuveitis and optic nerve swelling.
Another pattern seen less commonly is involvement of the choroid with focal or generalized breakdown of Bruch's membrane function with leakage on fluorescein angiography, corresponding to nodules or scars seen on retinal exam. This pattern may be confounded with lupus or other causes of multifocal choroiditis accompanied by aggressive intraocular inflammation: Both autoimmune and infectious etiologies would appear in this differential diagnosis.
Again, the clinical setting of this disease needs to be firmly kept in mind as the laboratory workup for other causes of choroiditis proceeds. ICG angiography reveals several patterns of choroidal circulatory disturbance in patients with Behçet's disease.35,36 These abnormalities are frequently missed on retinal exam and fluorescein angiography. The clinical significance of the ICG choroidal findings invites further study.
Treatment of ABD in its systemic and ocular manifestations consists of aggressive immunosuppression, usually with high-dose systemic steroid (intravenous and oral) initially and long-term immunosuppression with various disease modifying antirheumatic drugs. Antimetabolites, alkylating agents, and biologics, such as the anti-TNF-α agents, have been reported to control systemic and ocular involvement in ABD; it appears that controlling the disease systemically is valuable in saving retinal and choroidal tissue and circulation in the setting of ocular involvement.37 Local and intraocular steroids are also of value in controlling the accompanying uveitis and limiting its accompanying retinal and optic nerve damage.
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Since the mid-1980s, there has been growing global awareness of an important vasculopathy initially identified in patients with lupus but now known to occur as a primary entity in itself, called the antiphospholipid antibody syndrome (APS).
The current criteria for the clinical and laboratory diagnosis of the syndrome are the Sydney 2005 modifications of the 1999 Sapporo criteria. They include: arterial or venous thrombosis in any vascular territory, recurrent pregnancy loss or fetal growth restriction, occurring in close temporal relationship to (a) a positive IgG/M anticardiolipin antibody test, (b) a prolonged activated partial thromboplastin time (most commonly available worldwide), or (c) a dilute Russell viper venom time (a more specialized test less affected by anemia or pregnancy state).38 The Sydney modification adds a positive anti-β2 glycoprotein 1 antibody test and also addresses associated common findings, such as livedo reticularis and thrombocytopenia, which have not yet been validated worldwide. The entire April issue of the journal Lupus contains recent updates on all aspects of etiology, classification, pathophysiology, diagnosis and treatment from the Proceedings of the 13th International Symposium on Antiphospholipid Antibodies.
The spectrum of ocular involvement in APS is wide, ranging from amaurosis fugax and transient ischemic attacks to retinal hemorrhages, cotton wool spots, CRAO, CRVO, AION and ophthalmic artery39 and cilioretinal artery40 occlusions. As the occurrence of ocular and fundus manifestations in APS patients ranges from 15% to 88%,41,42 the diagnosis of APS must be considered in all patients with recurrent systemic or ocular thrombosis in the absence of known associated comorbidities associated with thrombophilia or those ocular thrombosis patients who have APS-related features, such as thrombocytopenia, migraine, transient ischemic attacks, or early strokes.43
More serious retinal problems involving large and small vessel occlusions of both arterioles and venules (see example in Figure 3), with rapid and profound loss of vision in both eyes, occur in the presence of antiphospholipid antibodies.44 Miserocchi et al. have shown in a small series of 13 patients with ocular findings and APS that 60% of patients had retinal vasculitis, and 7% had central retinal artery occlusions.45 These antibodies may occur in conjunction with lupus, with another connective-tissue disease, with an infection, or in a primary form of APS without an associated etiology, which is the most common occurrence.
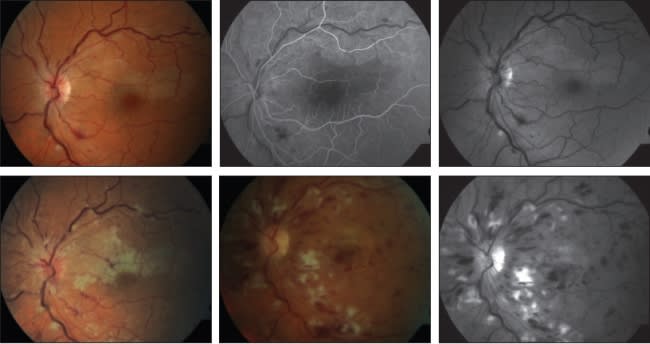
Figure 3. Images from a 54-year-old female with antiphospholipid antibody syndrome. Pretreatment color fundus photo of the right eye (top left) and midphase angiogram of the left eye (top center), and red-free fundus photo of the left eye (top right) show occlusions of both branch retinal artery and central retinal vein in the right eye. One week after the top row images were taken, a color fundus photo of the left eye (bottom left) shows a worsening condition. One month after the top row photos, fundus photos (color, bottom center, and red-free, bottom right) of the left eye show occlusive disease still progressing.
As with lupus, most patients with antiphospholipid antibodies alone have few if any retinal findings and have a good visual prognosis. A series of papers describes severe occlusive vasculopathy affecting the retina,46,47 choroid,48 and optic nerve49 of one or both eyes in patients with primary or secondary APS. An uncommon form of choroidal involvement in APS can mimic serpiginous choroidopathy.50
Concurrent central nervous system disease occurs in nearly 50% of patients with occlusive vascular involvement of the retina.44 The vascular changes noted in the retina occur in between 3.3% and 28.1% of patients with APS; it is likely that higher incidence is found in patients more severely affected by the disease systemically. In both lupus and APS, cotton wool spots alone represent a benign finding that resolves and is not usually a cause of visual loss. These areas of ischemia are associated with immune complex deposition in small vessels; they may respond to systemic immunosuppression.44
The characteristic ischemic vasculopathy noted in APS involves both arterial and venous trees, affecting large and small vessels and capillaries and leaving large areas of nonperfused retina in its wake.51 Retinal and optic nerve neovascularization occurs in up to 40% of eyes after vascular occlusions resulting in preretinal52 and vitreous hemorrhages,53 traction retinal detachments and significant loss of vision.44,54
Antiplatelet and anticoagulation therapy is of some benefit in limiting the spread of retinal ischemia; the underlying mechanism is recurrent thrombosis involving large and small vessels as a consequence of the effects of the antiphospholipid antibodies on coagulation and endothelial cells. Direct inflammatory damage to vessels or immune complex deposition plays a limited role in the pathogenesis of this form of retinal ischemia; immunomodulators have proved of little benefit in addressing and limiting retinal ischemia, except where coexisting intraocular inflammation occurs.55 Panretinal photocoagulation and vitrectomy are both valuable in managing the effects of the neovascularization. Use of intravitreal vascular endothelial growth factor inhibitors will also play a role in managing the effects of retinal vascular thrombosis.
Choroidal infarctions in one or both eyes have also been reported, along with Elschnig spots56 and multiple retinal pigment epithelium window defects seen on fluorescein angiography.57 The mechanism of vascular damage in the choroid is also likely to be thrombotic rather than inflammatory in nature. Similar retinal and choroidal changes have also been observed in children as young as six years old58 with primary APS, who lack many associated comorbidities such as hypertension, smoking or hypercholesterolemia. Central serous chorioretinopathy has also been observed in association with APS.56
In evaluating patients who have elevated levels of antiphospholipid or anticardiolipin antibodies, whether primary or secondary, the ophthalmologist also needs to assess the role of other diseases in producing ocular and retinal pathology. These diseases include hypertension, diabetes, and other comorbidities, such as renal failure, atherosclerosis, smoking or intercurrent infections, all of which cause endotheliopathy. The retinal findings common to APS, including retinal ischemia, vasculitis, large and small vessel occlusive disease, and retinal neovascularization, are also features of these inflammatory and vascular diseases. Unfortunately, the visual prognosis of this type of retinal vasculopathy is poor, with visual loss occurring in 80% of patients and with visual acuity of 20/60 or worse in 40% of patients.44 The visual outcomes worsen in the presence of retinal neovascularization and vitreous hemorrhage.
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is a chronic, multisystem inflammatory disease marked by the production of autoantibodies that circulate and damage blood vessels by multiple immune mechanisms: direct deposition on endothelial cells, immune complex formation and deposition, and multilayer lymphocyte-macrophage interactions.
Because blood vessels go everywhere, the criteria for the diagnosis of SLE recently updated by the American College of Rheumatology are protean: malar/discoid/photosensitive rashes, oral ulcers, arthritis, serositis, and renal/hematologic/neurologic disorders. These occur in association with positive serologies: antinuclear antibody test by indirect immunofluorescence on HEp-2 cells, a positive anti–double-stranded DNA test, a positive anti-Smith antibody, and positive antiphospholipid antibodies, most commonly IgG and IgM anticardiolipin, or a positive "lupus anticoagulant" test.59
Although worldwide in distribution, with differing frequency across ethnicities, SLE is estimated to occur at the rate of one in 250 patients, and a recent survey commissioned by the Lupus Foundation of America has estimated there are two million patients in the United States, with a 9:1 female-to-male preponderance. Genetically, lupus is a disease of multiplex loci leading to a hyperactive immune system, with amplification by environmental factors.60 Multiple autoantibodies may be present for a mean of seven years before clinically significant disease occurs, which is why full and early evaluation is needed, to prevent accumulated vascular damage.61
Ocular involvement in SLE is common, with abnormal retinal findings occurring in 30% of patients.63-65 Retinal involvement may be mediated by damage to small arterioles, capillaries and venules, with resulting ischemia involving the surrounding retina. Since 1999, optic neuropathy, when monocular, has been considered by some to represent a focal neuro-ophthalmic disease due to ciliary vascular thrombosis; conversely, the often bilateral optic neuropathy in SLE may be considered to be a more general neurologic disease due to multiple immunologic mechanisms, such as vasculitis.
Bilateral optic neuropathy has been thought to occur more frequently than unilateral disease, with an associated transverse myelitis.62 Arteriolar attenuation, cotton wool spots, microaneurysms, neovascularization of retinal vessels, and venous engorgement are indicators of this form of vascular insult.
Direct immune complex–mediated damage and microscopic occlusions of small vessels resulting from circulating autoantibodies are likely mechanisms for the destruction of vessels seen in the retina. In addition to directly impeding flow through small vessels, these complexes also activate complement and other immune effector/amplification systems, leading to further damage to vessels and the surrounding retina.66
A more recently proposed mechanism for retinal pathology occurring in lupus patients is that of an autoimmune retinopathy, similar to that seen in melanoma or other cancer patients: in this model, a variety of autoantigens destroy different cell types or layers of the retina, depending on their concentration, specificity, and access to retinal tissue through damaged blood vessels.67 While this idea is provocative, and the stimulation of the immune system by the presence of "altered self-antigens" as a driving force is thought to be operative in many autoimmune diseases, it remains unproven for ocular vasculopathy; cocontributory reasons for retinal circulatory changes in SLE include hypertension, azotemia, hyperviscosity and intercurrent infections.
Choroidal involvement in the eyes of lupus patients has been reported, based on ophthalmoscopic, fluorescein angiographic and ICG evaluations.65,68,69 Choroidopathy with multifocal inflammatory lesions, bone spicules with scarring, serous retinal detachments mimicking multifocal central serous retinopathy (see example in Figure 4), or large serous detachments or uveal effusions have been described.
As confusion can result as to whether ocular findings represent the effects of concurrent systemic steroid or intercurrent infections as confounders in an immunocompromised host, prompt diagnosis and appropriate treatment of the underlying lupus vasculopathy is crucial.
Systemic steroids and antimalarials (hydroxychloroquine preferentially, then chloroquine if needed), while commonly used mainstays for general SLE and organ-specific treatment, can also cause retinal abnormalities, albeit rare with the preferential use of hydroxychloroquine and careful longitudinal patient comanagement between rheumatologists and ophthalmologists. Currently, hydroxychloroquine is a recommended baseline therapy for all forms of lupus. The appearance of serous retinal detachments may indicate either active inflammatory disease or a side effect of systemic steroid therapy.70 Antimalarial retinal toxicity may result in permanent loss of vision.
Classically, a loss of central vision is noted 10 years or more after starting on the medication with visual field changes, retinal thinning around the macula, and a "bull's eye" pattern of retinal pigment epithelium scarring noted.71 Newer diagnostic methods, such as the use of spectral domain optical coherence tomography (SD-OCT), may identify changes in retinal architecture that occur before clinical symptoms and signs and that are more widespread than previously thought.72,73 Risk factors for antimalarial toxicity include high dose, long duration, high body mass index, age greater than 60, hepatic or renal disease, and concurrent retinal pathology. SD-OCT offers the promise of picking up retinal toxicity in a "preclinical" stage, before irreversible loss of vision occurs.64,72,73
The treatment of most lupus retinal and choroidal involvement is straightforward: rapid and aggressive initiation of systemic steroid and long-term immunosuppressive medication to control the underlying disease process.63,64 Careful attention to comorbidities, such as blood pressure, elevated blood glucose, and hyperlipidemia, is also critical to prevent unnecessary loss of retinal function and vision.
CONCLUSION
An appreciation of the systemic and retinal findings, and appropriate systemic and ocular therapy, of these vasculitic and vasculopathic diseases are important for the consulting ophthalmologist. Making a correct diagnosis among a multitude of possible etiologies of vision loss and retinal abnormalities affords us the opportunity to save a patient's life, as well as sight. Ultimately, initiating a thorough and complete medical, rheumatologic and ocular evaluation and paying close attention to the patient's complex medical issues enable the treating ophthalmologist to guide appropriate treatment systemically and for the eyes. RP
REFERENCES
1. Aristodemou P, Stanford M. Therapy insight: The recognition and treatment of retinal manifestations of systemic vasculitis. Nat Clin Pract Rheumatol. 2006;2:443-451.
2. Henkind P, Charles NS, Pearson J. Histopathology of ischemic optic neuropathy. Am J Ophthalmol. 1970;69:78-90.
3. Hayreh SS. Blood supply of the optic nerve head and its role in optic atrophy, glaucoma, and oedema of the optic disc. Br J Ophthalmol. 1969;53:721-748.
4. Gonzalez-Gay MA. Giant cell arteritis and polymyalgia rheumatica: two different but often overlapping conditions. Semin Arthritis Rheum. 2003;33:289-293.
5. Castro C, Gourley M. Diagnostic testing and interpretation of tests for autoimmunity. J Allergy Clin Immunol. 2010;125(Suppl 2):S238-S247.
6. Lopez-Diaz ML, Llorca J, Gonzalez-Juanatey C, Perla-Sagredo JL, Martin J, Gonzalez-Gay MA. The ESR is associated with the development of visual complications in biopsy-proven giant cell arteritis. Semin Arth Rheum. 2008;38:116-123.
7. McLeod D, Oji EP, Kohner EM, Marshall J. Fundus signs in temporal arteritis. Br J Ophthalmol. 1978;62:591-594.
8. Hayreh SS, Zimmerman MB. Fundus changes in central retinal artery occlusion. Retina. 2007;27:276-289.
9. Danesh-Meyer H, Savino PJ, Gamble GG. Poor visual prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology. 2005;112:1098-1103.
10. Nemec P, Jurecka T, Zampachová V, Masková Z, Soucek M. [Giant cell arteritis manifested by bilateral arteritis Anterior Ischemic Optic Neuropathy (AION)]. Vnitr Lek. 2008;54:1195-205.
11. Yoeruek E, Szurman P, Tatar O, Weckerle P, Wilhelm H. Anterior ischemic optic neuropathy due to giant cell arteritis with normal inflammatory markers. Graefes Arch Clin Exp Ophthalmol. 2008;246:913-915.
12. Galasso JM, Jay VM. An occult case of giant cell arteritis presenting with combined anterior ischemic optic neuropathy and cilioretinal artery occlusion. Semin Ophthalmol. 2004;19:75-77.
13. Daudin JB, Bluwol E, Chaine G, Rohart C. [Cotton-wool spots as first ocular manifestation of giant cell arteritis]. J Fr Ophtalmol. 2006;29:e28.
14. Asensio Sánchez VM, Pegalajar Maeso M, Rodr�guez Bravo I, Carrasco E. [Cotton-wool spots as the first manifestation of giant cell arteritis: two cases]. Arch Soc Esp Oftalmol. 2004;79:449-451.
15. Hayreh SS, Podhajshky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125:509-520.
16. Hayreh SS. Posterior ischaemic optic neuropathy: clinical features, pathogenesis, and management. Eye (Lond). 2004;18:1188-1206.
17. Schmidt D. Ocular ischemia syndrome-a malignant course of giant cell arteritis. Eur J Med. Res. 2005;10:233-242.
18. Hwang JM, Girkin CA, Perry JD, Lai JC, Miller NR, Hellman DB. Bilateral ocular ischemic syndrome secondary to giant cell arteritis progressing despite corticosteroid treatment. Am J Ophthalmol. 1999;127:102-104.
19. Moschos MM, Guex-Crosier Y. Giant cell arteritis: A rare cause of posterior vasculitis. Clin Ophthalmol. 2009;3:111-115.
20. Rajesh C, Cole M. Panuveitis as a presenting feature of giant cell arteritis. Br J Ophthalmol, 2000;84:337.
21. Valmaggia C, Speiser P, Bischoff P, Niederberger H. Indocyanine green versus fluorescein angiography in the differential diagnosis of arteritic and nonarteritic anterior ischemic optic neuropathy. Retina. 1999;19:131-134.
22. Coupal DJ, Patel AD. Siegrist streaks in giant cell arteritis. J Neuro-Ophthalmol. 2003;23:272-273.
23. Shah VA, Randhawa S, Boldt HC, Lee AG. Central serous chorioretinopathy in giant cell arteritis. Semin Ophthalmol. 2006;21:45-48.
24. Liozon E, Gondran G, Ly K, Loustaud V, Vidal E. Duration of treatment after eye involvement in giant cell arteritis. J Rheumatol. 2008;35:1220-1222.
25. Durrani K, Papaliodis GN. The genetics of Adamantiades-Behçet's disease. Semin Ophthalmol. 2008;23:73-79.
26. International Study Group for Behçet's Disease. Criteria for diagnosis of Behçet's disease. Lancet. 1990;335:1078-1080.
27. Ehrlich GE. Vasculitis in Behçet's disease. Int Rev Immunol. 1997;14:81-88.
28. Yamauchi Y, Cruz JM, Kaplan HJ, Goto H, Sakai J, Usui M. Suspected simultaneous bilateral anterior ischemic optic neuropathy in a patient with Behçet's disease. Ocul Immunol Inflamm. 2005;13:317-325.
29. Shima S, Nishimura K, Yamanaka K, et al. A case of Adamantiades-Behçet disease with ischemic optic neuritis (posterior optic neuropathy). J Dtsch Dermatol Ges. 2007;5:1010-1014.
30. Chan RV, Lee TC, Chaganti RK, Cestari DM, Kim MT, Lee S. Macular star associated with Behçet disease. Retina. 2006;26:468-470.
31. Ozdemir H, Mudun B, Karacorlu M, Karacorlu S. Serous detachment of macula in Behçet disease. Retina. 2005;25:361-362.
32. Ozdal PC, Ortaç S, Taşkintuna I, Firat E. Posterior segment involvement in ocular Behçet's disease. Eur J Ophthalmol. 2002;12:424-431.
33. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Kir N, Urgancioglu M. Neovascularization of the optic disc in Behçets disease. Jpn J Ophthalmol. 2006;50: 256-265.
34. Atmaca LS, Batioğlu F, Idil A. Retinal and disc neovascularization in Behçet's disease and efficacy of laser photocoagulation. Graefes Arch Clin Exp Ophthalmol. 1996;234:94-99.
35. Bozzoni-Pantaleoni F, Gharbiya M, Pirraglia MP, Accorinti M, Pivetti-Pezzi P. Indocyanine green angiographic findings in Behçet's disease. Retina. 2001;21:230-236.
36. Atmaca LS, Sonmez PA. Fluorescein and indocyanine green angiography findings in Behçet's disease. Br J Ophthalmol. 2003;87:1466-1468.
37. Kacmaz RO, Kempen JH, Newcomb C, et al.; Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study Group. Ocular inflammation in Behçet disease: incidence of ocular complications and of loss of visual acuity. Am J Ophthalmol. 2008;146:828-836.
38. Miyakis S, Lockshin MD, Atsumi T, et al. International concensus statement on an update of the classification criteria for definite antiphospholipid antibody syndrome. J Thromb Haemos. 2006;4:295-306.
39. Coroi M, Bontas E, Defransci M, Bartos D, Dorobantu M. Ocular manifestations of APS-minor features? Oftalmologia. 2007;51:16-22.
40. Carrero JL, Sanjurjo FJ. Bilateral cilioretinal artery occlusion in antiphospholipid syndrome. Retina. 2006;26:104-106.
41. Cobo-Soriano R, Sanchez-Ramon S, Aparicio MJ, et al. Antiphospholipid antibodies and retinal thrombosis in patients without risk factors: a prospective case-control study. Am J Ophthalmol. 1999;128:725-732.
42. Ostanek L, Modrzejewska M, Bobrowska-Snarska D, Brzosko M. [Ocular manifestations in patients with systemic lupus erythematosus and antiphospholipid syndrome]. Pol Arch Med Wewn. 2007;117(Suppl):65-69.
43. Yehuda D, Shoenfeld, Y, Toubi E. Looking into the eyes of patients with APS. Clin Rev Allergy Immunol. 2007;32:192-197.
44. Au A, O'Day J. Review of severe vaso-occlusive retinopathy in systemic lupus erythematosus and the antiphospholipid syndrome: associations, visual outcomes, complications and treatment. Clin Exp Ophthalmol. 2004;32:87-100.
45. Miserocchi E, Baltatzis S, Foster CS. Ocular features associated with aCLs: a descriptive study. Am J Ophthalmol. 2001;131:451-456.
46. Beckhauser AP, Arana LA, Larocca ST. Antiphospholipid syndrome causing bilateral occlusion of arteries and central retinal vein: case report. Arq Bras Oftalmol. 2008;71:282-285.
47. Chang PC, Chen WS, Lin HM, Chen SJ. Combined central retinal artery and central retinal vein occlusion in a patient with systemic lupus erythematosus and antiphospholipid syndrome. Lupus. 2010;19:206-209.
48. Ang LP, Yap EY, Fam HB. Bilateral choroidal infarction in a patient with antiphospholipid antibody: a case report. Clin Exp Ophthalmol. 2000;28:326-328.
49. Tsironi E, Gatselis N, Kotoula MG, et al. Ocular disorders as the prevailing manifestations of antiphospholipid syndrome: a case series. Cases J. 2009;2:159.
50. Tang J, Fillmore G, Nussenblatt RB. Antiphospholipid antibody syndrome mimicking serpiginous choroidopathy. Ocul Immunol Inflamm. 2009;17:278-281.
51. Suvajac G, Stojanovich L, Milenkovich S. Ocular manifestations in antiphospholipid syndrome. Autoimmun Rev. 2007;6:409-414.
52. Karagiannis D, Gregor Z. Valsalva retinopathy associated with idiopathic thrombocytopenic purpura and positive antiphospholipid antibodies. Eye (Lond). 2006;20:1447-1449.
53. Monshizadeh R, Werner M, Richards H, Tabandeh H, Bhatti MT. Vitreous hemorrhage as the presenting sign of antiphospholipid syndrome. Can J Ophthalmol. 2003;38:607-609.
54. Dori D, Gelfand YA, Brenner B, Miller B. Cilioretinal artery occlusion: an ocular complication of primary antiphospholipid antibody syndrome. Retina. 1997;17:555-557.
55. Cabrita FVL, Foster CS. Anticardiolipin antibodies and ocular disease. Ocul Immunol Inflamm. 2005;13:265-270.
56. Bolling JP, Brown JC. The antiphospholipid antibody syndrome. Curr Opin Ophthalmol, 2000;11:211-213.
57. Demirci FY, Kucukkaya R, Akarcay K, et al. Ocular involvement in primary antiphospholipid syndrome. Int Ophthalmol. 1998;22:323-329.
58. Hartnett ME, Laposata M, Van Cott E. Antiphospholipid antibody syndrome in a six-year-old female patient. Am J Ophthalmol. 2008;135:542-544.
59. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of SLE. Arthritis Rheum. 1997;40:1725.
60. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929-939.
61. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526-1533.
62. Giorgi D, Balacco-Gabrielli C. Optic neuropathy in SLE and APS: clinical features, pathogenesis, review of the literature, and proposed ophthalmological criteria for the diagnosis of APS. Clin Rheum. 1999;18:124-131.
63. Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm. 2004;12:87-99.
64. Davies JB, Rao PK. Ocular manifestations of systemic lupus erythematosus. Curr Opin Ophthalmol. 2008;19:512-518.
65. Nag TC, Wadhwa S. Vascular changes of the retina and choroid in systemic lupus erythematosus. Curr Neurovasc Res. 2006;3:159-168.
66. Sahu DK. An usual presentation of lupus retinopathy. Indian J Ophthalmol. 2008;56:72-73.
67. Cao X, Bishop RJ, Forooghian F, Cho Y, Fariss RN, Chan CC. Autoimmune retinopathy in systemic lupus erythematosus: histopathologic feature. Open Ophthalmol J. 2009;3:20-25.
68. Dhingra S, Stavrou P. Indocyanine green angiography in systemic lupus erythematosus-associated uveitis. Ocul Immunol Inflamm. 2004;32:69-73.
69. Gharbiya M, Pecci G, Baglio V, Gargiulo A, Allievi F, Balacco-Gabrieli C. Indocyanine green angiographic findings for patients with systemic lupus erythematosus nephropathy. Retina. 2006;26:159-164.
70. Barbón García JJ, Alvarez Suárez ML, Alvarez Navascués R, Viña Escalar C, Guerediaga Madariaga J. [Serous retinal detachment in systemic erithematosus lupus during corticosteroid therapy]. Arch Soc Esp Oftalmol. 2004;79:177-180.
71. Sivaraj RR, Durrani OM, Denniston AK, Murray PI, Gordon C. Ocular manifestations of systemic lupus erythematosus. Rheumatology. 2007;46:1757-1762.
72. Stepien KE, Han DP, Schell J, Godara P, Rha J, Carroll J. Spectral-domain optical coherence tomography and adaptive optics may detect hydroxychloroquine retinal toxicity before symptomatic vision loss. Trans Am Ophthalmol Soc. 2009;107:28-33.
73. Turgut B, Turkcuoglu P, Serdar Koca S, Aydemir O. Detection of the regression on hydroxychloroquine retinopathy in optical coherence tomography. Clin Rheumatol. 2009;28:607-609.
| Thomas E. Flynn, MD, was assistant professor of ophthalmology at the Harkness Institute of Columbia University in New York. Gale A. McCarty, MD, FACP, FACR, heads the Center for Lupus and APS at the Center for Rheumatology, Ocular Immunology, and Inflammation with Dr. Flynn at Ellsworth Uveitis and Retina Care in Maine. Neither author has any financial interests in any products mentioned in this article. Dr. Flynn can be reached via e-mail at tf86@columbia.edu. |