







PEER REVIEWED
Molecular Mechanisms of Neovascularization in AMD
MARK E. KLEINMAN, MD · JAYAKRISHNA AMBATI, MD
In the past 30 years, major discoveries have shed light on the molecular mechanisms that drive neovascular age-related macular degeneration (AMD) and choroidal neovascularization (CNV), ushering in a new paradigm of targeted therapeutics. As this highly specialized field of vasomolecular medicine continues to grow, an array of critical factors is being pursued as potential targets to halt and reverse the progression of neovascular AMD.
PATHOLOGIC NEOVASCULARIZATION IN AMD
Approximately 80% of patients suffering from AMD exhibit the "dry" atrophic form, whereas only 10% to 20% advance to the neovascular or "wet" form. Still, the latter is responsible for most vision loss in patients with AMD, a condition precipitated by the deleterious effects of CNV on retinal function. The critical molecular switches that activate the transition from dry to wet AMD are still under investigation.
Currently, a leading hypothesis suggests that the accumulation of inflammatory debris below the retinal pigment epithelium (RPE) and within the choroid leads to the production of proangiogenic cytokines and eventual invasion by pathologic vasculature.1 As many scientists and clinicians suspected, drusen, which usually precede the neovascular disease process, harbor numerous components capable of inducing oxidative injury, inflammation, and vasoproliferation, potentially triggering CNV formation. Out of a panel of recently discovered and promising therapeutic targets, the most efficacious CNV treatments have relied on the inhibition of vascular endothelial growth factor (VEGF), a dominant mediator of neovascularization.
| Mark E. Kleinman, MD, is a senior postdoctoral fellow in retinal cell biology at the University of Kentucky. Jayakrishna Ambati, MD, is professor and vice chair in the Department of Ophthalmology and Visual Sciences at the University of Kentucky. Dr. Kleinman reports no financial interests in this article. Dr. Ambati reports: minimal financial interests in Pfizer and Quark Pharmaceuticals; and moderate financial interests in Allergan. Furthermore, the University of Kentucky has filed patent applications on some of the technologies mentioned in the article. Dr. Ambati can be reached via e-mail at jamba2@email.uky.edu. |
THE DISCOVERY OF VASCULAR ENDOTHELIAL GROWTH FACTOR
A critical growth factor hypothesized as the "Factor X" in angiogenesis some 60 years ago was only isolated and characterized as VEGF in 1989.2-4 VEGF belongs to the highly conserved platelet-derived growth factor (PDGF) family, along with several other related proteins that constitute the VEGF subfamily, including placental growth factor (PlGF), VEGF-B, VEGF-C, and VEGF-D. For nomenclature purposes, the original VEGF protein was designated VEGF-A. Multiple isoforms of VEGF-A exist due to alternative splicing of the VEGF-A gene; however, the 165 amino acid variant in humans (VEGF-A165) is the most potent and prevalent among the major isoforms expressed during pathologic neovascularization.5
VEGF-A IS A CRITICAL TARGET IN THE PATHOGENESIS IN NEOVASCULAR AMD
The potential roles for VEGF-A signaling in the pathophysiology of CNV have become the focus of numerous translational investigations (Figure 1). CNV membranes surgically excised from patients with AMD were strongly reactive with anti-VEGF-A antibody,6,7 and pharmacological inhibition of VEGF-A decreased experimental laser-induced CNV in animal studies.8,9 These data ushered in an era of rapid development of VEGF-A-targeted therapies for use in patients with neovascular AMD, a boom that has already seen 3 major compounds come into clinical use: pegaptanib sodium (Macugen, OSI/Pfizer); bevacizumab (Avastin, Genentech); and ranibizumab (Lucentis, Genentech).
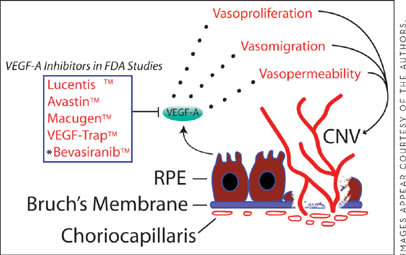
Figure 1. VEGF-A is a major contributor to the progression of wet AMD. During this process, abnormal choroidal blood vessels migrate through Bruch's membrane into the RPE and neural retina. Multiple VEGF-A targeted therapies are currently in advanced clinical trials for CNV, with some already gaining FDA approval and widespread use. Asterisk denotes therapies that target VEGF-A through RNA interference.
Pegaptanib is a modified 28-nucleotide RNA aptamer that binds to and inhibits VEGF-A165 and VEGF-A189. Inhibition of these isoforms was sufficient to significantly suppress experimentally induced retinal neovascularization in an animal model.10 However, advanced clinical trials with pegaptanib in patients with CNV only reduced the vision loss compared to placebo at 1- and 2-year endpoints. The sole VEGF-A-targeted therapeutic agent approved by the US Food and Drug Administration that has been shown to improve vision in patients with CNV is ranibizumab, which has thus become a widely used treatment for the condition. Ranibizumab is a neutralizing VEGF-A antibody fragment that is capable of inhibiting all known isoforms of VEGF-A. In pivotal clinical trials for the treatment of classic and occult CNV, ranibizumab demonstrated superior visual outcomes compared to sham-treated eyes, as well as those receiving photodynamic therapy. The results of these large clinical studies bolster the importance of VEGF-A as a critical provascular factor in the pathogenesis of CNV. Further major clinical trials of ranibizumab are under way to optimize its dosing frequency and potential use in combination therapy.
Another VEGF-A-targeted therapeutic modality called RNA interference is also in advanced clinical trials for neovascular AMD. These drugs are based on the recently discovered molecule, short interfering RNA (siRNA), a 21-25 nucleotide double-stranded RNA that can specifically target messenger RNA (mRNA) for degradation. This gene-silencing mechanism works through an enzyme complex called RNA-induced silencing complex, which separates siRNA, anneals it to a targeted complementary mRNA, and then cleaves the duplexed RNA. There are 2 clinical investigations of VEGF-targeting, siRNA-based drugs for CNV: bevasiranib (OPKO Health), which targets VEGF-A mRNA and is in phase 3 clinical trials; and Sirna-027/AGN211745 (Allergan), which targets one of the VEGF-A receptors, VEGFR-1, and is currently in a phase 2 clinical trial. Although this pharmaceutical approach holds great promise, we recently discovered striking evidence that all siRNAs activate the immune system through a cell surface receptor called toll-like receptor 3 (TLR3), an event that resulted in neovascular suppression independent of the siRNA sequence or target.11 Further evidence confirming generic siRNA binding to TLR3 has recently appeared.12
Furthermore, it was observed that if these drugs are forcibly internalized into cells and allowed to silence their intended target in the absence of TLR3 activation, only the siRNA directed against VEGF-A caused a reduction in experimental laser-induced CNV.11 This finding supports previous work that VEGFR-1 is not a bona fide proangiogenic receptor. More significantly, patients receiving these siRNA-based drugs may be at risk for unanticipated vascular and immune-related side effects due to TLR3 activation.
Many other important components within the VEGF family may contribute to neovascular AMD. The expression of genes important in VEGF regulation and activation, such as the coreceptors neuropilin-1 and -2, as well as PlGF, were upregulated in affected eyes.13-16 There may be further therapeutic potential by inhibiting some of these other VEGF-related factors, a question that could be partially answered in phase 3 CNV trials with VEGF-Trap-Eye (Regeneron), a drug that binds PlGF and VEGF-B, in addition to all VEGF-A isoforms.
CONTRADICTORY ROLES OF VEGF-A SIGNALING IN NEOVASCULAR AMD
Although VEGF-A is clearly a major contributor to CNV, the diverse roles of this proangiogenic cytokine in its pathogenesis remain unclear. This is evident from clinical trials of the currently available VEGF-A inhibitors. In studies with pegaptanib, there was an inverse dose response with visual outcome, and while the lowest dose decreased the rate of vision loss, it did not prevent an increase in CNV lesion size (52% for pegaptanib vs 57% for sham over 1 year).17 Even with ranibizumab, the CNV lesion continued to grow despite treatment or was, at best, unchanged. After 2 years of treatment with ranibizumab, some of the lesions exhibited progression of atrophy, a finding attributed to an independent advancement of dry AMD and not as a side effect.
Clinicians and patients must realize that the long-term effects of continual anti-VEGF-A therapy are unknown. Genetically altered mice that do not have VEGF-A isoforms in the RPE demonstrated profound abnormalities, including photoreceptor and choriocapillary atrophy.18,19 With pharmacologic blockade of VEGF-A, mice exhibited increased death of retinal neurons following experimental injury.20 Collectively, these findings demand that clinicians and scientists continue to work toward a more complete understanding of the molecular biology of VEGF-A in CNV, especially given that many patients with neovascular AMD are now receiving VEGF-A inhibitors.
CELLULAR RESPONSES TO VEGF-A RECEPTOR BINDING
As a multifaceted cytokine, VEGF-A influences several endothelial cell pathways resulting in growth, survival, and vascular permeability. Although its signal transduction is mediated primarily through 2 receptor tyrosine kinases, VEGFR-1 and VEGFR-2, the latter is implicated as the principal proangiogenic transducer. Normal human eyes and surgically excised CNV membranes are known to express VEGFR-1 and VEGFR-2.21 Controversially, the role of VEGFR-1 has been characterized as a decoy receptor evolved to trap free VEGF-A, thereby preventing continuous VEGFR-2 activation. In multiple models of pathologic angiogenesis, VEGFR-1 has functioned dichotomously as it both amplifies22,23 and antagonizes24 neovascularization; however, it does not appear that VEGFR-1 does this by simply serving as a decoy. On the contrary, cell-based studies demonstrated that VEGFR-1 can repress VEGFR-2-mediated endothelial cell proliferation through active signaling.25-27 We found that VEGFR-1 transduction by VEGF-A can suppress VEGFR-2 activation in the laser-induced model of CNV.28 Thus, VEGF-A may be able to act in both pro- and antiangiogenic capacities.
REGULATION OF ANGIOGENESIS BY THE EXTRACELLULAR MATRIX
It is now understood that the biologic activities of VEGF-A and other cytokines are influenced by the interaction of cells with the extracellular matrix (ECM). ECM proteins channel cell-ECM communication and greatly influence tissue remodeling events, including angiogenesis. For almost 20 years, it has been known that CNV membranes contain an abundance of ECM proteins similar to those found in granulation tissue during wound healing. Surgically excised CNV tissue contained fibrin and fibronectin,29 both of which are important in ECM-guided angiogenesis. Integrin receptors, which regulate cell signaling through interaction with various components of the ECM, are also important in CNV formation30 and have been successfully targeted in animal models of CNV.31,32
The pathogenesis of CNV requires invasion of pre-existing ECM and the formation of new avenues of vasculature. During this process, specific enzymes are expressed that degrade the basement membranes, allowing for subsequent vascular ingrowth (Figure 2). Several years ago, a link was discovered between proangiogenic factors found in CNV membranes and the upregulation of specific matrix-metalloproteinases (MMP-2 and MMP-9), which are capable of ECM breakdown.33-35 Tissue inhibitors of MMPs, which prevent dissolution of ECM by MMPs, are also expressed in CNV membranes and have provided a novel therapeutic target.36,37 Another proteinase mechanism known to be involved in CNV progression is the urokinase plasminogen activator pathway, which is also being evaluated as an alternative treatment modality.38-40
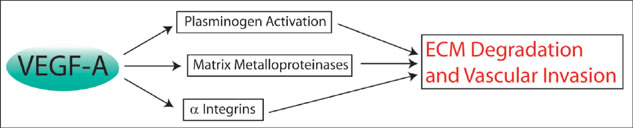
Figure 2. VEGF-A interacts with a host of proteins that are capable of degrading the ECM and thus compromising the integrity of Bruch's membrane. Thinning and eventual fracture of the membrane allow the abnormally growing choroid blood vessels to invade the retinal layers.
More novel ECM proteins are being discovered that may be involved in VEGF-A signaling and CNV pathogenesis. The matricellular secreted protein, acidic and rich in cysteine (SPARC) is induced by VEGF-A and affects remodeling, cellular migration, and angiogenesis.41,42 SPARC decreased the stimulatory activity of VEGF-A on endothelial cell proliferation,43 but at this time, the exact mechanism of interaction between SPARC and VEGF-A is unknown. In our investigations, we were surprised to find that intravitreous VEGF-A suppressed CNV when injected after laser injury. Mouse eyes treated with VEGF-A prior to laser injury exhibited the typical proangiogenic response and enhanced CNV. Further experiments suggest that this unexpected biphasic VEGF-A activity may be due to decreased SPARC levels during the acute phase of laser-induced injury. Investigations are under way to delineate the critical factors in this paradoxical mechanism. These data may help improve the scheduling and dosing of VEGF-A-targeted therapeutics in order to restrict pharmacologic effects to the intended action of suppressing and reversing CNV. In addition, the restriction of SPARC expression to the macula in primates may help explain the macular predilection of CNV in humans.
MOLECULAR SIGNALS OF CELLULAR INVASION IN NEOVASCULAR AMD
Many molecular mediators pertinent to CNV progression, including VEGF-A, are also known to result in the recruitment of inflammatory and stem/progenitor cells to injured tissues.44,45 Similar to epithelial wound healing, there appears to be an orchestrated arrival of various inflammatory cell types in CNV lesions that dictate the course of disease. From other wound-healing studies, it is known that inflammatory cell recruitment is directed by the expression of cytokines, such as monocyte chemoattractant protein-1 (Ccl2; also known as MCP-1), GRO (also known as CXCL1), and macrophage inflammatory protein-1 and -2 (MIP-1, MIP-2), some of which have been found in CNV tissues and may be contributory to its progression.
It is also believed that stem/progenitor cells are recruited to sites of CNV and incorporate into pathologic vasculature. Animal studies have now demonstrated that interfering with stem/progenitor cell homing to the sites of CNV through the modulation of the cytokine stromal-derived factor-1 and its receptor, CXCR4, inhibits neovascular growth,46,47 a finding that offers another potential treatment for the human condition.
OTHER PROANGIOGENIC FACTORS INVOLVED IN NEOVASCULAR AMD
Several other molecular factors have been implicated in the development of CNV through descriptive expression analyses in human specimens, including transforming growth factor-beta 1 (TGF-β1), PDGF, fibroblast growth factor-2, insulin-like growth factor-1 and estrogen. Endogenous antiangiogenic molecules have also been identified, such as pigment-epithelium-derived factor and thrombospondin-1, that are decreased in CNV lesions48,49 and are capable of regulating retinal and choroidal neovascularization in animal studies.50,51 These factors provide yet another level of control in this increasingly complex system of vascular regulation.
NEOVASCULAR PROGRESSION VIA COMPLEMENT ACTIVATION
Many of the currently identified drusen components are potential inflammatory mediators and may directly upregulate cytokines that are critical for disease progression to neovascular AMD. From this perspective, it is proposed that drusen do not simply precede the pathogenic growth of choroidal blood vessels, but actually promote it. Recent data have directly linked complement components 3a (C3a) and 5a (C5a), known components of AMD drusen, to the upregulation of VEGF-A (Figure 3).52 RPE cell cultures treated with C3a or C5a secreted significantly more VEGF-A, whereas choroidal endothelial cells did not. In animal studies, VEGF-A concentrations in the RPE and choroid peaked just 4 hours after intravitreous C3a/C5a administration. This well-localized and rapid response suggests that the RPE, and perhaps other resident cells, plays a major role in VEGF-A production after complement activation. These results may also help explain the abrupt switch-like progression to neovascular AMD in patients with previously dry fundus exams.
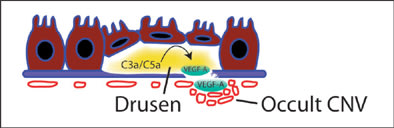
Figure 3. Complement factors are abundant in drusen and lead to the activation of numerous inflammatory pathways. C3a/C5a upregulates RPE production of VEGF-A which is able to stimulate choroidal endothelial cells to proliferate, migrate, and form leaky vasculature beneath Bruch's membrane, as in occult CNV, or break through to sub-RPE space and neural retina layers, as in classical CNV.
CONCLUSION
As further molecular research is conducted on the pathobiology of neovascular AMD, more mechanisms will be revealed, thus providing additional therapeutic targets for this devastating disease. Already, we are within reach of a detailed map of critical proteins and cells that mediate the pathogenesis of CNV, a feat that has taken more than 20 years of dedicated science. The wide use of VEGF-A-specific therapies and their proven efficacy demonstrates the incredible power of this modernized approach to pharmacologic design and solidifies the utility of the current animal models designed to mimic the human form of neovascular AMD. With these novel molecular tools and predictive models, future scientific investigations into this disease process will continue to yield invaluable insight with the hope of offering clinicians the information necessary to improve the patients' outcomes. RP
REFERENCES
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705-732.
- Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971;133:275-288.
- Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851-858.
- Gospodarowicz D, Abraham JA, Schilling J. Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary-derived folliculo stellate cells. Proc Natl Acad Sci USA. 1989;86:7311-7315.
- Usui T, Ishida S, Yamashiro K, et al. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368-374.
- Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855-868.
- Kvanta A, Algvere PV, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929-1934.
- Saishin Y, Takahashi K, Silva RL, et al. VEGF-TRAPR1R2 suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195:241-248.
- Krzystolik MG, Afshari MA, Adamis AP, et al. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch Ophthalmol. 2002;120:338-346.
- Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198:483-489.
- Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591-597.
- Pirher N, Ivičak K, Pohar J, Benčina M, Jerala R. A second binding site for double-stranded RNA in TLR3 and consequences for interferon activation. Nat Struct Mol Biol. 2008 Jun 22 [Epub ahead of print].
- Martin G, Schlunck G, Hansen LL, Agostini HT. Differential expression of angioregulatory factors in normal and CNV-derived human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2004;242:321-326.
- Lim JI, Spee C, Hangai M, et al. Neuropilin-1 expression by endothelial cells and retinal pigment epithelial cells in choroidal neovascular membranes. Am J Ophthalmol. 2005;140:1044-1050.
- Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165. J Biol Chem. 2000;275:18040-18045.
- Rakic JM, Lambert V, Devy L, et al. Placental growth factor, a member of the VEGF family, contributes to the development of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3186-3193.
- Gragoudas ES, Adamis AP, Cunningham ET, Jr., Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805-2816.
- Marneros AG, Fan J, Yokoyama Y, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167:1451-1459.
- Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado A, D'Amore PA. Soluble VEGF isoforms are required for the maintenance of the retinal pigment epithelium (RPE)-choriocapillaris complex in the adult. Invest Ophthalmol Vis Sci. 2008;49:1537.
- Shima DT, Nishijima K, Jo N, Adamis AP. VEGF-mediated neuroprotection in ischemic retina. Invest Ophthalmol Vis Sci. 2004;45:3270-.
- Kim I, Ryan AM, Rohan R, et al. Constitutive expression of VEGF, VEGFR-1, and VEGFR-2 in normal eyes. Invest Ophthalmol Vis Sci. 1999;40:2115-2121.
- Carmeliet P, Moons L, Luttun A, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575-583.
- Hiratsuka S, Maru Y, Okada A, Seiki M, Noda T, Shibuya M. Involvement of flt-1 tyrosine kinase (vascular endothelial growth factor receptor-1) in pathological angiogenesis. Cancer Res. 2001;61:1207-1213.
- Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993-1008.
- Rahimi N, Dayanir V, Lashkari K. Receptor chimeras indicate that the vascular endothelial growth factor receptor-1 (VEGFR-1) modulates mitogenic activity of VEGFR-2 in endothelial cells. J Biol Chem. 2000;275:16986-16992.
- Zeng H, Dvorak HF, Mukhopadhyay D. Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) receptor-1 down-modulates VPF/VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent pathways. J Biol Chem. 2001;276:26969-26979.
- Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993-1008.
- Nozaki M, Sakurai E, Raisler BJ, et al. Loss of SPARC-mediated VEGFR-1 suppression after injury reveals a novel antiangiogenic activity of VEGF-A. J Clin Invest. 2006;116:422-429.
- Grossniklaus HE, Martinez JA, Brown VB, et al. Immunohistochemical and histochemical properties of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1992;114:464-472.
- Friedlander M, Theesfeld CL, Sugita M, et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc Natl Acad Sci USA. 1996;93:9764-9769.
- Kamizuru H, Kimura H, Yasukawa T, Tabata Y, Honda Y, Ogura Y. Monoclonal antibody-mediated drug targeting to choroidal neovascularization in the rat. Invest Ophthalmol Vis Sci. 2001;42:2664-2672.
- Fu Y, Ponce ML, Thill M, Yuan P, Wang NS, Csaky KG. Angiogenesis inhibition and choroidal neovascularization suppression by sustained delivery of an integrin antagonist, EMD478761. Invest Ophthalmol Vis Sci. 2007;48:5184-5190.
- Steen B, Sejersen S, Berglin L, Seregard S, Kvanta A. Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1998;39:2194-2200.
- Hoffmann S, He S, Ehren M, Ryan SJ, Wiedemann P, Hinton DR. MMP-2 and MMP-9 secretion by RPE is stimulated by angiogenic molecules found in choroidal neovascular membranes. Retina. 2006;26:454-461.
- Das A, Fanslow W, Cerretti D, Warren E, Talarico N, McGuire P. Angiopoietin/Tek interactions regulate MMP-9 expression and retinal neovascularization. Lab Invest. 2003;83:1637-1645.
- Das A, McLamore A, Song W, McGuire PG. Retinal neovascularization is suppressed with a matrix metalloproteinase inhibitor. Arch Ophthalmol. 1999;117:498-503.
- Della NG, Campochiaro PA, Zack DJ. Localization of TIMP-3 mRNA expression to the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1996;37:1921-1924.
- Rakic JM, Lambert V, Munaut C, et al. Mice without uPA, tPA, or plasminogen genes are resistant to experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:1732-1739.
- Das A, McGuire P. Role of urokinase inhibitors in choroidal neovascularization. Semin Ophthalmol. 2006;21:23-27.
- Lambert V, Munaut C, Carmeliet P, et al. Dose-dependent modulation of choroidal neovascularization by plasminogen activator inhibitor type I: Implications for clinical trials. Invest Ophthalmol Vis Sci. 2003;44:2791-2797.
- Kato Y, Lewalle JM, Baba Y, et al. Induction of SPARC by VEGF in human vascular endothelial cells. Biochem Biophys Res Commun. 2001;287:422-426.
- Hasselaar P, Sage EH. SPARC antagonizes the effect of basic fibroblast growth factor on the migration of bovine aortic endothelial cells. J Cell Biochem. 1992;49:272-283.
- Kupprion C, Motamed K, Sage EH. SPARC (BM-40, osteonectin) inhibits the mitogenic effect of vascular endothelial growth factor on microvascular endothelial cells. J Biol Chem. 1998;273:29635-29640.
- Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. Embo J. 1999;18:3964-3972.
- Lemieux C, Maliba R, Favier J, Theoret JF, Merhi Y, Sirois MG. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood. 2005;105:1523-1530.
- Sengupta N, Caballero S, Mames RN, Timmers AM, Saban D, Grant MB. Preventing stem cell incorporation into choroidal neovascularization by targeting homing and attachment factors. Invest Ophthalmol Vis Sci. 2005;46:343-348.
- Lima E, Silva R, Shen J, et al. The SDF-1/CXCR4 ligand/receptor pair is an important contributor to several types of ocular neovascularization. FASEB J. 2007;21:3219-3230.
- Uno K, Bhutto IA, McLeod DS, Merges C, Lutty GA. Impaired expression of thrombospondin-1 in eyes with age related macular degeneration. Br J Ophthalmol. 2006;90:48-54.
- Bhutto IA, McLeod DS, Hasegawa T, et al. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82:99-110.
- Mori K, Duh E, Gehlbach P, et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253-263.
- Zamiri P, Masli S, Kitaichi N, Taylor AW, Streilein JW. Thrombospondin plays a vital role in the immune privilege of the eye. Invest Ophthalmol Vis Sci. 2005;46:908-919.
- Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. 2006;103:2328-2333.