







Inflammation and Age-related Macular Degeneration
New research expands understanding of how inflammation and the complement system contribute to AMD disease progression.
JAYAKRISHNA AMBATI, MD
Recent years have witnessed tremendous advances in the understanding of how inflammation and complement activation relate to the development and progression of age-related macular degeneration (AMD). Since 2005, there has been greater recognition of the role that the complement system plays in the formation of drusen and the progression of AMD. The complement system, or cascade, is orchestrated by 30 proteins that are involved in the body's defense against micro-organisms. Recently, abnormal or aberrant function in various genes or complement factors have been shown to dramatically increase the likelihood of the development and progression of AMD from the dry to the wet form. Continued research in this area and new genetic findings relating to the complement system will lead to the development of diagnostic and therapeutic treatments that can benefit a larger proportion of the early AMD population.
OVERVIEW
Age-related macular degeneration is the leading cause of irreversible vision loss among the elderly in industrialized nations.1,2 The bilateral ocular condition affects the central area of the retina, the macula, and it is grouped into 2 basic clinical categories: the "dry" atrophic form and the exudative neovascular, or "wet," form. Although the wet form represents only about 10% of AMD, it is responsible for about 90% of serious visual loss in AMD. This rapidly progressive vision loss is a result of pathologic new blood vessels, or choroidal neovascularization (CNV), invading the retina from the underlying choroid through fractures in Bruch's membrane, the extracellular matrix between the choroids, and the retinal pigmented epithelium (RPE).2,3
| Jayakrishna Ambati, MD, is professor and vice chair in the Department of Ophthalmology and Visual Sciences at the University of Kentucky in Lexington. Dr. Ambati is named as an inventor on patent applications filed by the University of Kentucky relating to technologies pertaining to the subject matter. |
There is no good preventive therapy or cure for AMD at this time. Approved treatment options for wet AMD include antineovascular agents, photodynamic therapy, and thermal laser photocoagulation. No current therapies exist for dry AMD, save for the Age-Related Eye Disease Study (AREDS) vitamin formulation, which benefits a small fraction of patients in this category. New therapies relating to inflammation and the complement system hold hope to treat patients prior to the development of CNV (Figure 1).
INFLAMMATION AND THE COMPLEMENT SYSTEM
The earliest clinical hallmark of AMD is the presence of drusen.1 While our understanding of molecular events presaging AMD has grown in recent years, its pathogenesis has remained puzzling and our therapeutic arsenal still is somewhat limited. However, new research and development continue to herald new potential interventions.
Drusen nearly always precede and increase the risk of CNV, the late vision-threatening stage of AMD. Although intraocular inflammation is not clinically apparent in AMD, there is now evidence that supports an influential role for inflammation in this condition. Recent studies of the molecular composition of drusen have pinpointed local inflammation as a key element in the pathogenesis of AMD.
In 2001, Hageman et al.4 proposed a unifying hypothesis of drusen biogenesis that attempted to incorporate a large body of structural, histochemical, and molecular data pertaining to drusen composition and development. Under this paradigm, they described an integrated ultrastructural, histochemical, molecular biological, and biochemical approach to identify specific molecular pathways associated with drusen biogenesis. They noted that a thorough understanding of the composition of drusen and sources of drusen-associated material was most likely to provide new insight into the pathobiology underlying AMD.
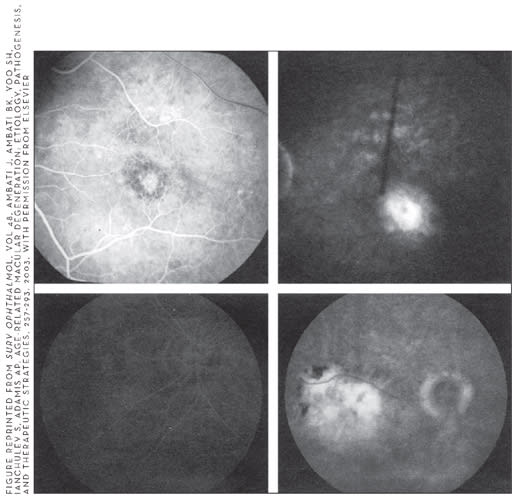
Figure 1. (top) Early and late phase fluorescein angiogram (FA) of classic CNV. Early phase shows a discrete area of RPE/choroidal leakage, which increases in the last phase. (bottom) Early and phase FA of occult CNV. Early phase shows no choroidal leakage, but late phase shows marked leakage at the level of the RPE/choroid of unknown origin.
Their study revealed that proteins associated with inflammation and immune-mediated processes are prevalent among drusen-associated constituents and transcripts that encode a number of these molecules were detected in retinal, RPE, and choroidal cells. These data also led to the observations that dendritic cells, potent antigen-presenting cells, are closely associated with drusen development and that complement activation is a key pathway that is active both within drusen and along the RPE-choroid interface.
Also in 2001, Penfold et al.5 looked at the etiological and immunological aspects of AMD. They provided historical descriptions of the involvement of inflammatory cells. Evidence implicating inflammation in the pathogenesis of AMD involving macrophages, giant cells, and microglia was derived from observations of human and animal subretinal neovascular lesions. The role of humoral factors, such as antiretinal autoantibodies and acute phase proteins, together with clinical observations were also surveyed. The researchers concluded that these data demonstrated the involvement of immunity in the pathogenesis of AMD. Though they noted that further investigation was needed to determine the degree to which immunity is causative or contributory in wet and dry AMD, they also commented that the use of anti-inflammatory agents to relieve the condition further indicated the existence of an inflammatory component.
Then, in 2002, Anderson et al.6 concluded that local inflammation plays a role in drusen biogenesis. Their findings suggested that, similar to the processes that occur in the other age-related diseases, accumulation of extracellular plaques and deposits elicits a local chronic inflammatory response that exacerbates the effects of primary pathogenic stimuli. In this study, these investigators examined the tissue specimens from one or both eyes of >400 human donors with light, confocal, or electron microscopy, together with antibodies to specific drusen-associated proteins, to help characterize the transitional events in drusen biogenesis. They quantified messenger RNA from the RPE and choroid of donor eyes was used to determine whether local ocular sources for drusen-associated molecules exist.
The results indicated that debris derived from degenerate RPE cells become sequestered between the RPE basal lamina and Bruch's membrane. The researchers proposed that this debris constitutes a chronic inflammatory stimulus and a potential "nucleation" site for drusen formation. The entrapped debris then becomes the target of encapsulation by inflammatory mediators, some of which are contributed by the RPE and perhaps other local cell types and some of which are extravasated from the choroidal circulation.
LEUKOCYTE INVOLVEMENT
Recent research suggests that immune-mediated processes also play a key role in the biogenesis of drusen. Histochemical staining of drusen and basal laminar (BlamD) in Bruch's membrane indicates accumulation of neutral fats and phospholipids with age. It has been suggested that focal concentration of these materials may produce a powerful chemotactic stimulus for leukocytes. This may, in fact, occur via the complement cascade. It appears as if macrophages preferentially engulf the wide-banded collagen of BlamD in patients with AMD. In addition, further supporting the inflammatory paradigm is the presence of circulating antiretinal autoantibodies in the sera of patients with AMD, which may or may not be an associated or reactive response.1
Leukocyte presence in histological studies of CNV has brought forth interest in the role of leukocytes in the development of neovascular AMD. In studies conducted over the last 2 decades, leukocytes, and more specifically macrophages, have been implicated in the pathogenesis of AMD in both human and animal models (Figure 2).
In 1993, van der Schaft et al.7 showed in eyes with CNV that Bruch's membrane thins for some area around the break. Beneath the thinned areas lay regions of deeper choroidal infiltration made up of macrophages, lymphocytes, fibroblasts, and myofibroblasts. Also, activated macrophages may play an active role rather than passive in nonspecific inflammation because these macrophages secrete proteolytic enzymes, such as collagenase and elastase. These enzymes can erode the thinned Bruch's membrane and facilitate migration of choroidal capillaries.
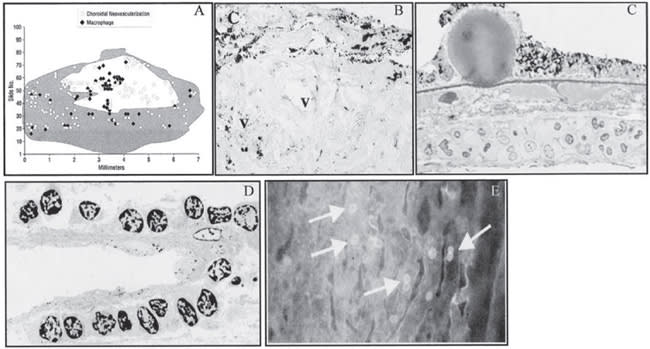
FIGURE 2A REPRINTED FROM ARCH OPHTHALMOL, VOL 118, GROSSNICKLAUS HE, CINGLE KA, YOON YD, ET AL. IMMUNOHISTOCHEMICAL AND HISTOCHEMICAL PROPERTIES OF SURGICALLY EXCISED SUBRETINAL NEOVASCULAR MEMBRANES IN AGE-RELATED MACULAR EDEMA, 464-472, 2000, WITH PERMISSION FROM ELSEVIER. FIGURE 2 REPRINTED FROM OH H, TAKAGI H, TAKAGI C, ET AL. THE POTENTIAL ANGIOGENIC ROLE OF MACROPHAGES IN THE FORMATION OF CHROIDAL NEOVASCULAR MEMBRANES. INVEST OPHTHALMOL VIS SCI. 1999;40:1891-1898. COURTESY OF INVESTIGATIVE OPHTHALMOLOGY AND VISUAL SCIENCE. FIGURES 2C AND 2D REPRINTED FROM PROGRESS IN RETINAL AND EYE RES, VOL. 20, PENFOLD PL, MADIGAN MC, GILLIES MC, PROVIS JM. IMMUNOLOGICAL AND AETIOLOGICAL ASPECTS OF MACULAR DEGENERATION, 385-414, 2001, WITH PERMISSION FROM ELSEVIER. FIGURE 2E IS REPRINTED FROM AM J PATHOL (2001), 158:1161-1172 WITH PERMISSION FROM THE AMERICAN SOCIETY OF INVESTIGATIVE PATHOLOGY.
Figure 2. A. Histologic 2-D reconstruction demonstrating macrophages in spatial proximity to CNV. The shaded area indicates and area of intact RPE and the unshaded area corresponds to RPE/defect disciform scar. B. Immunohistochemistry for CG68 reveals numerous macrophages located in the RPE monolayer, the stroma, and around the neovascular vessels in a CNV membrane caused by AMD. C. Histology of of eye of patient with dry AMD showing leukostasis in the congested choroid beneath hyaline druse compromising predominantly monocytes. D. Electron micrograph of eye of patient with CNV showing accumulation of leukocytes adherent to abluminal surface of a new subretinal blood vessel. E. Choriocapillaris of transgenic mouse overexpressing VEGF in the RPE showing with abundant adherent leukocytes (arrows).
In situ hybridization studies, such as those by Grossniklaus et al.8 and Spandau et al.,9 have documented high levels of monocyte chemoattractant protein-1 (MCP-1) in RPE cells of AMD eyes. This may account for the presence of macrophages in CNV. It is likely that under flow conditions, MCP-1 can induce leukocyte adhesion to vascular endothelium as shown by Gerszten et al.10 in 1999.
In addition, growing evidence exists that leukocytemediated angiogenesis involves the interaction of cellular adhesion molecules and vascular endothelial growth factor (VEGF).1 For example, in 2000, Miyamoto et al.11 and Radisavljevic et al.12 provided evidence that VEGF induces the expression of intercellular adhesion molecule-1 (ICAM-1) on tumor and retinal vascular endothelium and regulates leukocyte adhesion to endothelial cells.
The complexity of this system does not end there. Other studies reported in the literature show that the ICAM-1 blockade decreases VEGF-induced leukostasis in the retina and angiogenesis in the cornea. These systems are mutually involved. For instance, leukocytes possess receptors for and migrate in response to VEGF, which they can also produce and release. The modulation of angiotensin-2-induced leukocyte adhesion by reactive oxygen intermediates and nitric oxide also supports this complexity.1
We reported in 2003 that laser-induced CNV is markedly slowed in mice genetically deficient in ICAM-1 or its cognate receptor on leukocytes, CD18.13 This suggests a causal role for leukocyte adhesion to vascular endothelium in the CNV development. We investigated the role of the leukocyte adhesion molecules CD18 and ICAM-1 in the development of CNV. We used laser photocoagulation to induce CNV in wild-type C57BL/6J mice and species-specific counterparts with targeted homozygous disruption of the CD18 or ICAM-1 gene. Expression of CD18 and ICAM-1 after laser injury was assessed by immunostaining. CNV responses were compared on the basis of en masse volumetric measurements obtained by confocal microscopy 2 weeks after laser injury and by determination of fluorescein angiographic leakage at 1, 2, and 4 weeks after laser injury. The site of laser injury showed upregulation of ICAM-1 and invasion by CD18-positive leukocytes within 1 day of laser injury. Significantly fewer lesions exhibited fluorescein leakage defined as pathologically significant in CD18-deficient mice at weeks 1, 2, and 4 and in ICAM-1-deficient mice at 1 and 4 weeks, compared with the control. There were a greater number of lesions without fluorescein leakage in CD18-deficient mice than in the other 2 groups at all time points. The volume of CNV in CD18- and ICAM-1-deficient mice was significantly less than in wild type. Combined with the immunolocalization of ICAM-1 and MCP-1 in human CNV, and the presence of leukocytes concentrated around newly formed CNV, the evidence for leukocyte involvement in the development of CNV is compelling.
COMPLEMENT FACTORS
Increasing evidence suggests the complement system also plays a significant role on the pathogenesis of AMD. As such, this research is beginning to provide a more definitive picture of the relevant cellular events, genetic factors, and biochemical processes associated with early AMD.
Most recently, it has been shown that abnormal or aberrant function in various genes, or complement factors, can increase the likelihood of the development and progression of AMD from the dry to the wet form. Therefore, complement activation represents a step forward, with potential of being the first target for prophylactic intervention for AMD. VEGF-blockade therapy targets blood vessels after they have already grown, while the complement system provides a series of targets for prevention of CNV.
Gehrs et al.2 support this new paradigm of AMD pathogenesis that has come from research of the complement system activation and various complement factors, such as Complement Factor H (CFH). They report that the complement cascade is activated by an initiating event or trigger. The exposure to infection or some other triggering event in genetically susceptible individuals, coupled with impaired complement regulatory function, leads to the sustained activation of the complement cascade, drusen formation, and eventually the development of AMD.
Furthermore, the CFH gene, which encodes the major inhibitor of the complement alternative pathway, is the first gene identified in independent studies that confers a significant genetic risk for the development of AMD. The researchers noted that this finding, together with the subsequent identification of AMD-associated variants in the related complement genes BF and C2, provides compelling evidence that the innate immune system and, particularly, uncontrolled regulation of the alternative pathway of complement play a central role in the pathobiology of AMD.
In 2006, my colleagues and I conducted a study3 in which we found that both in patients with AMD and in a recently described mouse model of AMD, early sub-RPE deposition of complement components C3 and C5 occurs. This suggests a contributing role for these inflammatory proteins in the development of AMD. Our study demonstrated that bioactive fragments of these complement components C3a and C5a are present in drusen of patients with AMD, and that C3a and C5a induce VEGF expression in vitro and in vivo. Further, we demonstrated that C3a and C5a are generated early in the course of laser-induced CNV, an accelerated model of neovascular AMD driven by VEGF and recruitment of leukocytes into the choroid. We also showed that genetic ablation of receptors for C3a or C5a reduces VEGF expression, leukocyte recruitment, and CNV formation after laser injury and that antibody-mediated neutralization of C3a or C5a or pharmacological blockade of their receptors also reduces CNV.
Collectively, these findings establish a mechanistic basis for the clinical observation that drusen predispose to CNV, revealing a role for immunological phenomena in angiogenesis and providing therapeutic targets for AMD.
CONCLUSIONS
Based on this exciting research, the future of AMD is exceptionally bright. In the next several years, this research will lead to a more complete understanding of the genetic basis for many more AMD cases.
This new paradigm has already spurred the development of early diagnostics, novel bioassays, and new animal models based on this paradigm. There are now a dozen companies with various complement-targeting compounds that are in various stages of preclinical or clinical trial and eventually new therapies for modulating the alternative pathway of the complement "at risk" individuals prior to the onset of CNV or geographic atrophy will be developed.
In addition, this understanding of genetic information will lead to new genetic screening tests, similar to those in other disease areas which will identify individuals who are most at risk for developing AMD later in life. RP
REFERENCES
1. Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257-293.
2. Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration — emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450-471.
3. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328-2333.
4. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immunemediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705-732.
5. Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385-414.
6. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411-431.
7. van der Schaft TL, Mooy CM, de Bruijn WC, de Jong PT. Early stages of agerelated macular degeneration: an immunofluorescence and electron microscopy study. Br J Ophthalmol. 1993;77:657-661.
8. Grossniklaus HE, Ling JX, Wallace TM, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119-126.
9. Spandau U, Toksoy A, Holz F, Schrader W, Liesenhoff H. High expression of angiogenic chemokines in choroidal neovascularization membranes. Invest Ophthalmol Vis Sci. 2000;41:S836.
10. Gerszten RE, Garcia-Zepeda EA, Lim YC, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718-723.
11. Miyamoto K, Khosrof S, Bursell SE, et al. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am J Pathol. 2000;156:1733-1739.
12. Radisavljevic Z, Avraham H, Avraham S. Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J Biol Chem. 2000;275:20770-20774.
13. Sakurai E, Taguchi H, Anand A, et al. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:2743-2749.