







Laser and Pharmacologic Therapies in the Treatment of Diabetic Macular Edema
VICTOR H. GONZALEZ, MD
 INTRODUCTION
INTRODUCTION
Diabetic retinopathy is a major cause of blindness in Europe and North America,1 affecting persons in all phases of their working lives.2 Diabetic retinopathy (DR) has several levels of severity, ranging from a mild nonproliferative form characterized by the existence of microaneurysms, through more serious manifestations characterized by vascular abnormalities of increasing prominence.3 At any stage of DR, patients may also develop diabetic macular edema (DME), resulting from breakdown of the blood retinal barrier (BRB).3 The formal definition of DME established by the Early Treatment Diabetic Retinopathy Study (ETDRS)4 is thickening of the retina and/or the presence of hard exudates within 1 disc diameter of the center of the macula. This definition is further classified as clinically significant if at least
1 of the following conditions are met: retinal thickening within 500 μm of the center of the macula; hard exudates within 500 μm of the center of the macula if there is thickening of the adjacent retina; or a zone of retinal thickening 1 disc area in size providing some part is within 1 disc diameter of the center of the macula (Figure 1). Recently, a severity scale for DME has been proposed to facilitate patient screening. It includes two principal DME levels, absent and present, based on whether there is thickening or hard exudates in the posterior pole. Where present, DME is defined as mild, moderate, or severe, corresponding to whether these sequelae are distant, near, or involving the center of the macula.5
The risk of developing DME is substantial in individuals with diabetes mellitus. In a 10-year study, risk was found to be 20.1% in patients with type 1 diabetes, 25.4% in insulin-dependent type 2 disease, and 13.9% in type 2 patients not requiring insulin.6 In DME, leakage from hyperpermeable capillaries and microaneurysms are major contributors to vision loss.7 This review will discuss current practices in DME management, with a particular focus on novel approaches that have been developed as a result of deeper understanding of the underlying pathophysiology of this disease.
PATHOPHYSIOLOGY OF DR/DME
The pathophysiology of DR/DME is complex, involving changes in both the retinal vasculature and in retinal neurons. While abnormal neuronal function, as observed in the electroretinogram, is common8 and can be detected even before the onset of microvascular lesions,9-11 the focus of current treatment strategies is on the edema and neovascularization that accompany DR. DME-related disturbances of vision reflect the formation of leaky vessels, while closure of existing vasculature, followed by proliferative neovascularization and associated complications, further exacerbates the visual deficits. The initial changes in the retinal vasculature include the degeneration and loss of the pericytes enveloping the retinal capillaries, together with thickening of the capillary basement membrane, adherence by leukocytes to the endothelium, and capillary blockage. These blockages, in turn, promote the loss of endothelial cells and the formation of acellular vessels.12,13 The resultant nonperfusion and local hypoxia lead to increased local expression of promoters of angiogenesis14 such as vascular endothelial growth factor (VEGF), one of the principal molecular targets of new therapies for DME discussed in the following sections.
Several molecular mechanisms have been posited as candidates for mediating the link between hyperglycemia and vascular damage, including the accumulation of polyols, the formation of advanced glycation end products, the overall increase in oxidative stress with concomitant generation of free radicals, and the pathological activation of protein kinase C (PKC) (for reviews see Caldwell et al12; Sheetz and King15; and Fong et al16). It should also be noted that both reactive oxygen intermediates17 and advanced glycation end products18 act to induce the elevated expression of VEGF, another therapeutic target.
Finally, it should be noted that the vascular damage that accompanies diabetes bears many of the hallmarks of an inflammatory process.10 McLeod et al19 reported that intercellular adhesion molecule-1 (ICAM-1), which promotes leukocyte adhesion, was significantly elevated throughout the choroidal vasculature and in retinal blood vessels in diabetic patients. Moreover, diabetic patients showed increases in the numbers of neutrophils in the retina and choroid, suggesting that this increased neutrophil adhesion via ICAM-1 may contribute to the capillary blockage. In addition, vitreous levels of ICAM-1 are elevated in DME, while patients with DR also have elevated plasma levels of tumor necrosis factor-a (TNF-a), a key inflammatory cytokine.20
DIAGNOSIS
Medical history and fundus examination are the bases for diagnosis of DR. The standard method for assessing the severity of DR/DME was defined by the ETDRS and involves stereoscopic fundus photography of 7 standard fields through dilated pupils.21 Owing to its complexity, however, it is not employed routinely in the clinic. While ophthalmoscopy is the common clinical tool, its use by nonophthalmologists has relatively poor sensitivity compared with the ETDRS standard.22 One approach to simplifying the screening process is through the use of nonmydriatic retinal imaging platforms such as the Joslin Vision Network system (Boston), with electronic transmission to a central reading center for evaluation; this technique has shown a close match to determinations made by dilated retinal examinations in assessing the level of DR and DME.23 Fluorescein angiography, while able to detect early indications of DME through fluorescein leakage from capillaries, is primarily used to confirm clinical diagnosis, to stage retinopathy, and to plan treatment, rather than simply for screening, owing to its inherent invasiveness and expense.3 In addition, capillary leakage does not necessarily signal DME. Optical coherence tomography (OCT) is increasingly being employed to assess retinal thickness as a surrogate marker for efficacy in treating DME.24 Several groups have reported that OCT measurements of retinal thickness show significant correlations with visual acuity (VA) in patients with DME25-27; however, other studies have reported contradictory findings.28,29 Thus, the applicability of OCT in the management of patients with DME is still questionable. Finally, another noninvasive approach, the Heidelberg Retinal Tomograph, based on confocal laser imaging, has also shown promise as an assay system for macular edema.30
TREATMENT
Laser Ablation of Affected Areas
Current treatment approaches for DME are derived from the ETDRS; in patients with DME and mild to moderate nonproliferative DR, focal laser photocoagulation decreased the probability of a 15-letter loss in VA from 8% to 5% at 1 year and from 24% to 12% at 3 years.4 Benefit was restricted to patients whose DME was defined as clinically significant. While visual prognosis was worse for eyes with decreased VA at baseline, eyes with 20/40 VA were more likely to show an improvement of �6 letters in VA. Because the treatment is inherently destructive, it may entail side effects such as paracentral scotomas, alterations in color perception, choroidal neovascularization, metaplasia of the retinal pigment epithelium (RPE), and inadvertent burns to the center of the macula.7,12 Laser scars may also expand significantly after treatment,31 an issue of particular concern for burns close to the macula. Macular grid photocoagulation technique could theoretically reduce the risk of such complications by sparing the fovea region. However, a recent study presented at the American Academy of Ophthalmology (AAO) Subspecialty Day 2006 compared the modified ETDRS focal laser photocoagulation (m-ETDRS) with a mild macular grid (MMG) technique and suggested that m-ETDRS should continue to be the standard treatment for DME. A total of 323 study eyes with DME were randomized 1:1 to these 2 treatment options, and after a 1-year follow-up, there was a trend favoring
m-ETDRS group in both VA and anatomical (OCT) results.32
One approach to minimizing the retinal destruction is through the use of a micropulsed laser in which short bursts of laser are applied rather than a continuous burn. Friberg and Karatza,33 treating patients with macular edema secondary to branch vein occlusion or DR, reported that 26 of 34 (76%) newly treated patients and 10 of 15 (67%) previously treated patients experienced clinical improvement at 6 months. Similarly, Moorman and Hamilton34 reported that panretinal and grid pattern photocoagulation performed using the micropulse mode with the laser on for 100 to 300 μs and off for a cycle of 1700 to 1900 μs resulted in the resolution of macular edema at 6 months in 22 of 39 eyes (56%). Although laser therapy is the only proven treatment, it does not address the underlying pathogenic mechanism of the disease. In addition, some patients with DME may qualify for laser therapy as defined by ETDRS guidelines because of the diffuse nature of their edema but have poor outcomes.4,35 Pharmacological therapies provide us with an opportunity to treat this important group of patients.
Pharmacological Approaches
Pharmacological treatments for DME are premised on interference with the molecular processes that lead to the development of diabetic vascular damage, not only to reverse it after it has developed, but also, in the ideal case, to prevent its occurrence. These initiatives have led to several new therapies that have shown promise in clinical trials or case series, including drugs targeting VEGF, PKC, and TNF-a, as well as the use of an anti-inflammatory corticosteroid.
Inhibition of VEGF

VEGF is the most potent of the known factors promoting both physiological and pathological angiogenesis (for a review, see Ferrara36). Splicing of the human VEGF gene leads to 6 principal isoforms containing 121, 145, 165, 183, 189, and 206 amino acids;36,37 the corresponding rodent isoforms are 1 amino acid shorter. VEGF165, the most extensively studied isoform, exists both in a soluble form as well as bound to the extracellular matrix; in contrast, VEGF121 lacks the heparin-binding domain and is not matrix-bound, while isoforms larger than VEGF165 are highly basic and exist primarily in association with the matrix.36 VEGF is a ligand for 2 receptor tyrosine kinases, VEGFR-1 and VEGFR-2, that act through downstream signaling cascades.38 VEGF acts through many mechanisms in promoting angiogenesis (Table 1).39-49 A concerted research effort over the last decade has established that VEGF is both necessary and sufficient for the neovascularization that is characteristic of ocular diseases such as age-related macular degeneration (AMD) and DR (for a review, see Ng and Adamis50). For example, intravitreal injection of VEGF into eyes of nondiabetic monkeys leads to the development of many of the changes typically seen in diabetes, including the development of microaneurysms and tortuous, leaky vessels.51,52
It should be noted, however, that VEGF acts in a wide variety of physiological processes, some related to its importance for angiogenesis and others not; these latter include bone growth,53,54 female reproductive cycling,53,55 kidney development and tissue maintenance,56,57 wound healing,58,59 skeletal muscle regeneration,60 maintenance of the health of hepatic cells,61 vasorelaxation,62 and survival of a wide variety of neuronal cell types,63,64 including retinal neurons.65 VEGF plays an ongoing role in trophic maintenance of capillaries in a variety of organs66; in the eye it is essential for the development of the choriocapillaris,67 a tissue that receives continuing trophic support through VEGF secretion by the RPE.68 Thus, therapeutic strategies based on the inactivation of VEGF must take heed of potential adverse events. For example, intravenous administration of bevacizumab (Avastin, Genentech), an antibody that inactivates all VEGF isoforms indicated for the treatment of colorectal cancer, has been accompanied by increased incidences of hypertension, bleeding, gastrointestinal perforations, and thromboembolic events.69-72 For ocular diseases, these concerns are reduced in some measure when intravitreally administered,73 but even here caution may be warranted since there is continuity between the vitreous and plasma compartments, especially in the context of diabetes, where the BRB is compromised (for a discussion, see van Wijngaarden et al74). While these are theoretical concerns, no clear evidence has been generated in the clinical trials using selective vs nonselective VEGF-blocking strategies. Continued surveillance will help address these questions.
Several properties of VEGF are of importance for the particular pathophysiology of DME. First, the expression of VEGF is upregulated by hypoxia,75,76 which, in turn, is a local consequence of capillary dropout. In the retina, VEGF is expressed by many cell types, including pericytes, endothelial cells, glial cells, neurons, and the RPE,68, 77,78 and upregulation of VEGF by hypoxia is common to all.68,77,79 Second, VEGF is the most potent known promoter of
vascular permeability, some 50 000 times greater than
histamine80; mechanisms underlying this enhanced permeability include the induction of fenestrations in the endothelial cell membrane,81 disruption of tight junctions through phosphorylation of their constituent proteins,82 and the development of caveolae, which may result in the formation of transendothelial pores83 (for reviews, see Olsson et al38 and Weis and Cheresh84).
There is also good evidence that VEGF plays a key role in mediating the inflammatory changes that are characteristic of DR/DME. In an early study, VEGF was found to be elevated in the vitreous of patients with DR.85 These elevations were also accompanied by increases in ICAM-1 levels.86 Furthermore, investigations into the the development of BRB breakdown in a rodent model of diabetes provided strong evidence that retinal vascular damage is mediated by increases in VEGF. This induces upregulation of ICAM-1, leading to retinal leukostasis and endothelial cell damage through Fas-FasL-mediated apoptosis.87-91
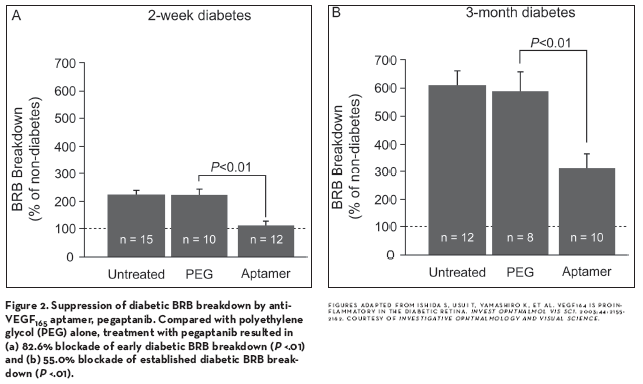
Detailed studies of the involvement of VEGF indicate that one isoform, VEGF165 (or the rodent counterpart 164), may play an especially important role in both diabetes-induced retinal damage, as well as in the hypoxia-related neovascularization that can be a manifestation of DR. Compared to VEGF120, intravitreal injection of VEGF164 in nondiabetic rats led to approximately twice the level of ICAM-1 upregulation, retinal leukostasis, and BRB breakdown. In addition, intravitreal injection of pegaptanib sodium (Macugen, [OSI] Eyetech/Pfizer), a polyethylene-glycolated RNA aptamer that binds to VEGF164 but not to VEGF120, was able to reverse these effects in diabetic animals, leading to significant restoration of the BRB (Figure 2).92
In a mouse model of retinopathy of prematurity, pathological ischemic neovascularization was accompanied by a dramatic increase in the upregulation of VEGF164 compared to VEGF120, and intravitreal injection of pegaptanib inhibited this pathological neovascularization while sparing normal physiological retinal neovascularization.93 In contrast, injection of a VEGFR-Fc fusion protein that inactivates all VEGF isoforms inhibited both physiological and pathological neovascularization (Figure 3).93 Additional studies determined that VEGF164 was dispensable in protecting retinal neurons against cell death under conditions of ischemia.65 Together with its actions in reversing BRB breakdown, this selectivity for pathological neovascularization suggested that intravitreal pegaptanib might provide a safe treatment for reducing the excess permeability of DME, as well as the neovascularization that can be a feature of DR.
SELECTIVE AND NONSELECTIVE ANTAGONISTS OF VEGF
Pegaptanib sodium
Pegaptanib sodium, administered intravitreally, has shown clinical efficacy against all angiographic subtypes of drs and �15 letters of VA (34% vs 10%, P=.003, and 18% vs 7%, P=.12, respectively). Moreover, proportions of patients with absolute decreases in retinal thickness of
�75 μm and �100 μm also favored 0.3 mg pegaptanib over sham (49% vs 19%, P=.008, and 42% vs 16%, P=.02, respectively).98 Pegaptanib was well tolerated at all doses. One case of endophthalmitis was reported; a total of
652 injections were administered.
In a post hoc analysis of a subset of 16 patients with documented retinal neovascularization at study entry, 8 of the 13 (62%) receiving pegaptanib showed evidence of regression on fundus photographs at 36 weeks. In contrast, none oflkawbhklfbalskdbfajksbdvklubam,snbvbasm,dnbvklajbwembfm,ZBx,bvm,bzS The experimental design included an intravitreal injection of 0.3 mg pegaptanib every 6 weeks for 30 weeks (for a total of 6 injections), with results compared to patients (or fellow eyes) receiving panretinal photocoagulation. Of 10 eyes receiving pegaptanib, 9 showed total regression after 3 weeks with the remaining eye showing partial regression; regression at 6 weeks was 89%. Of
8 eyes receiving photocoagulation, 7 remained active at
6 weeks. Mean changes in VA at 6 weeks were a gain of
4.9 and of 6.5 letters in the pegaptanib and photocoagulation groups, respectively, while only the pegaptanib-treated group experienced a reduction in foveal thickness.
Ranibizumab
Ranibizumab (Lucentis, Genentech) is a fragment antigen binding (Fab) fragment, which, like bevacizumab, reactsrthrthrthrthrthrth with all VEGF isoforms (for a review, see Ferrara et M101). Ranibizumab has shown efficacy in phase 3 trials as an intravitreal treatment for AMD.102,103 More recently, it has been examined as a treatment for DME in open-label pilot-scale studies, r4porting the outcomes of 10 patients. Nguyen et al104 administered 5 injections of 0.5 mg ranibizumab over 6 months and reported significant improvements in mean VA (20/80 to 20/40) and reductions in macular volume and retinal thickness; no ocular or systemic events were observed. In the second study, Chun et al105 administered
3 injections of either 0.3 mg or 0.5 mg ranibizumab over
2 months. At 3 months, 4 patients had gained �15 letters, gained �10 letters, while the mean retinal thickness decrease was 45.3 ±196.3 μm for the 0.3-mg group, and 197.8 ±85.9 μm for the 0.5-mg group. While there were no systemic adverse events, 5 cases of mild to moderate ocular inflammation were reported.
Bevacizumab
There is considerable interest in the use of bevacizumab as an intravitreally administered agent, especially as it may bring considerable cost advantages if efficacy and safety can be demonstrated. Although no studies evaluating bevacizumab in the management of DME have been published to date, data were recently presented at the 2006 AAO meeting. A multicountry retrospective review of the outcomes of 82 eyes following a 1.25 mg dose of bevacizumab found a significant improvement in mean BCVA and central macular thickness on OCT (P=.0001 for each outcome) at an average of 12.7 ±4.9 weeks.106 No ocular or systemic adverse events were observed. With respect to DR, there have been several small studies showing success in achieving regression of DR-induced ocular
neovascularization.107,108
Avery et al109 have reported on the use of intravitreal bevacizumab in treating 45 eyes of 32 patients with retinal and/or iris revascularization. Intravitreal doses ranging from 6.2 μg to 1.25 mg were given. All patients (44/44 eyes) had complete or partial reduction in leakage, as assessed by fluorescein angiography, within 1 week of injection. Leakage of iris neovascularization completely resolved in 9 of
11 eyes (82%), while complete resolution of leakage of neovascularization of the disc was achieved in 19 of 26 eyes (73%). In 2 cases, both involving doses of 1.25 mg, there was also a subtle decrease in neovascularization of the uninjected fellow eye, suggesting that significant systemic levels were achieved at this dose. This observation led to the use of the smaller doses, with therapeutic effects seen with as low a dose as 6.2 μg in the study eye. Recurrence of leakage was seen as early as 2 weeks postinjection, whereas in some cases none was observed throughout the 11 weeks of follow-up. The authors suggest that while the relatively rapid recurrence may ultimately prove limiting, this issue requires more detailed study and that in any event intravitreal bevacizumab may provide a useful adjunctive therapy together with panretinal photocoagulation and surgical approaches.
Inhibition of PKC-b
Protein kinase Cs are a family of enzymes, some of which have been found to be activated in diabetes owing to elevated levels of diacyl glycerol, advanced glycation end products, and reactive oxygen intermediates (for a review, see Sheetz and King15), as well as VEGF.110 PKC-b2 activation has in turn been linked to mechanisms mediating leukostasis and vascular damage. Ruboxistaurin is an orally administered inhibitor specific for PKC-b2 in rats; both orally and intravitreally administered ruboxistaurin significantly inhibited increased retinal permeability induced by intravitreally administered VEGF.111 In rats with streptozotocin-induced diabetes mellitus, ruboxistaurin administered intravitreally restored the retinal circulatory parameters to values approximating those of nondiabetic animals,112 while intravitreal injection of another PKC inhibitor, GF109203X, was shown to reverse BRB breakdown, resulting in retinal permeability essentially identical to that of control animals.113
Several recent clinical trials examining the use of ruboxistaurin have now reported varying results in treating diabetes-related retinal complications. In a multicenter, double-masked, randomized, placebo-controlled study involving 252 patients with moderately severe to very severe nonproliferative DR, orally administered daily doses ranging from 8 mg to 32 mg or placebo were given for up to 46 months. While the drug was well tolerated, it did not prevent the progression of DR, although the 32-mg dose did provide a significant benefit in reducing the risk of moderate vision loss and also reduced the risk of sustained moderate vision loss from 25% to 10% in eyes with significant macular edema at baseline.114 Qualitatively similar results were seen in an 18-month trial involving 41 patients with macular edema.115 In a 4-week study examining retinal blood flow parameters, diabetes-associated impacts on retinal blood flow and retinal circulation time were ameliorated by ruboxistaurin treatment.116
In a 3-year, randomized, double-masked, placebo-controlled trial involving 686 patients to assess the impact of ruboxistaurin treatment (32 mg/day) on visual loss associated with moderately severe to very severe nonproliferative DR, the drug reduced the incidence of sustained moderate visual loss from 9.1% in the placebo group to 5.5% in the ruboxistaurin group (P=.034).117 For patients with baseline clinically significant macular edema >100 μm from the center of the macula, ruboxistaurin significantly reduced the probability of progression to within the 100-μm zone
(68% vs 50%; P=.003); in addition, initial laser treatment for DME was reduced from 37.9% in the control group to 28.0% in those receiving the drug (P=.008).117 Rubox-
istaurin was well tolerated with no evidence of increased incidence of adverse events linked to the drug. It should be noted, however, that despite these encouraging results, the Food and Drug Administration has recently requested that a new phase 3 trial be performed before ruboxistaurin can be approved as a treatment for DR.118,119
Modulators of inflammation
K Triamcinolone acetonide. The inflammatory nature of the retinal vascular pathology and neovascularization in DR/DME suggests that standard anti-inflammatory agents such as corticosteroids could be of use as therapies. The most commonly employed is the synthetic corticosteroid triamcinolone acetonide. In preclinical studies triamcinolone inhibited cytokine-induced upregulation of ICAM-1 by endothelial cells120 and reduced hypoxia-induced upregulation of VEGF by cultured RPE cells.121 In other studies, triamcinolone inhibited preretinal and optic nerve head neovascularization following experimentally induced retinal vein occlusion in pigs.122
Off-label triamcinolone has been used investigationally for a variety of ocular diseases, including DME and AMD (for a review, see Jonas123). Principal adverse events include ocular hypertension in approximately 40% of eyes, medically uncontrollable ocular hypertension in 1% to 2% of eyes, and development of cataracts (predominantly subcapsular) in approximately 15% to 20% of patients.123 The high risk of adverse events may preclude the use of triamcinolone as monotherapy for conditions requiring long-term therapy.124
Recently, the results of prospective, placebo-controlled, randomized studies were reported using triamcinolone for treating DME. Jonas et al125 randomized 28 eyes to receive a single intravitreal injection of 20 mg triamcinolone while
12 eyes received placebo injections. At 6 months, VA was increased significantly in the triamcinolone group vs the control group, with 11 of 23 (48%) of the triamcinolone-treated group having at least a 2-line improvement compared to none of the controls. In a 2-year, double-masked study involving eyes with DME that persisted or recurred following laser treatment, 69 eyes of 43 patients were randomized to receive 4 mg intravitreal triamcinolone or placebo (subconjunctival saline).126 Eyes with a loss of at least 5 letters in VA from the previous visit and persistent central macular thickness exceeding 250 μm were retreated with triamcinolone, with a minimum of 6 months required between repeat treatments. At 2 years, VA improvement of �5 letters was seen in 19 of 34 (56%) eyes receiving triamcinolone compared with 9 of 35 (26%) control eyes (P=.006); the authors suggested that improvement among controls may have reflected intensified efforts by patients in the trial to control their hyperglycemic risk factors. Glaucoma medication was required in 15 of 34 (44%) treated eyes compared to 1 of 30 (3%) of controls (P=.0002) with 2 of the treated eyes requiring trabeculectomy; cataract surgery was performed in 15 of 28 (54%) vs none of 21 control eyes.
Finally, it should be noted that implantable devices are being developed to permit long-term release of steroids in the vitreous while obviating the need for repeated injections. These include an intravitreal implant for the delivery of fluocinolone for treatment of noninfectious uveitis in the posterior segment and indications such as DME,127 as well as a biodegradable implant for the intravitreal delivery of dexamethasone to treat various ocular ischemic diseases.128 Although the long-acting fluocinolone implant showed promising results in improving visual acuity and decreasing retinal thickness in patients with DME at both 1 and 2 years after initiating therapy, nearly all phakic eyes develop cataracts within 2 years of use and over 30% of patients require a surgical procedure to control intraocular pressure (IOP). Fluocinolone acetonide implant is currently approved for patients with uveitis only. A biodegradable, implantable extended-release product that delivers dexamethasone directly to the posterior segment has been evaluated in a phase 2 clinical trial in
306 patients with DME. Patients administered 700 μg had a significant improvement in VA of �2 or more lines on the ETDRS chart when compared with patients who did not receive the implant (P=0.02). IOP increased to
�25 mm Hg at some point in 32 eyes but was controlled with topical antihypotensive medications. Phase 3 trials are currently underway.129
K Infliximab. Infliximab is a chimeric monoclonal antibody against TNF-a, a major inflammatory cytokine (for a review, see Siddiqui and Scott130) that is elevated in the plasma of diabetic patients relative to controls.20 Studies in a rat model of diabetes showed that inactivation of TNF-a by a soluble TNF-a-receptor/Fc protein could reduce leukocyte adhesion and BRB breakdown.90 The effects of TNF-a in potentiating leukocyte adhesion to retinal endothelial cells appear to be mediated in part by PKC-b2-dependent phosphorylation of the glycosylating enzyme core 2 b 1,6-N-acetylglucosaminyltransferase.131 In addition, TNF-a can upregulate endothelial cell expression of VEGF132 and ICAM-1,110 providing yet another pathway for inducing leukocyte-mediated vascular damage.
The clinical evidence for efficacy of infliximab treatment is slim, based on only 1 case report using intravenous infusion of infliximab at monthly intervals at a dose of
5 mg/kg in 7 eyes of 4 patients with DME, 6 of which were refractory to laser photocoagulation. Within 1 month of treatment, macular thickness had decreased in 5 of
7 eyes with DME, with further reductions in macular thickness and improvements in VA reported at 2 months.133 Very similar positive results have been reported by this same group of investigators in treating AMD with intravenous infliximab.134 These preliminary findings suggest that TNF-a inhibition may be beneficial in treating ocular neovascular diseases. While the only clinical experience to date has involved systemic administration of infliximab, the advantages of lower overall doses and ready tissue access suggest that intravitreal injection of this agent, either alone or in combination with therapies such as pegaptanib, might be a useful strategy in treating DR/DME.
CONCLUSIONS
The recommendations of the ETDRS constituted a major advance in the treatment of DR and DME. These treatment options have now been extended by the research effort of the last decade into the pathophysiology of the disease. It should be noted that the ETDRS macular laser studies required 3 years to observe a clear benefit from the laser therapy, whereas the pharmacological approach appears to yield results within weeks after administration. The pathogenesis of DR involves many pathways, but the data appear to suggest that, in some cases, the macular edema is dependent on high VEGF levels.98 This may explain why some patients demonstrate a marked improvement of their edema with anti-VEGF therapy. Pharmacologic therapies may allow us to reduce our need for macular laser in select patients and perhaps employ more selective peripheral retinal ablation, instead of the nonselective approach we currently utilize with combination therapy. Current ongoing trials and those in development are needed to further define the best treatment protocols for this complicated group of patients. As with AMD, it appears that pharmacologic therapies for the management of DR/DME will cause a paradigm shift in how we care for these patients. RP
REFERENCES
1. Resnikoff S, Pascolini D, Etya'ale D, et al. Global data on visual impairment in the year 2002. Bull World Health Organ. 2004;82:844-851.
2. Kempen JH, O'Colmain BJ, Leske MC, et al, for the Eye Diseases Prevalence Research Group. The prevalence of diabetic retinopathy among adults in the United States. Arch Ophthalmol. 2004;122:552-563.
3. Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: pathophysiology, screening, and novel therapies. Diabetes Care. 2003;26:2653-2664.
4. Early Treatment Diabetic Retinopathy Study Research Group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Arch Ophthalmol. 1985;103:1796-1806.
5. Wilkinson CP, Ferris FL 3rd, Klein RE, et al. Global Diabetic Retinopathy Project Group. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003;110:1677-1682.
6. Klein R, Klein BE, Moss SE, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. XV. The long-term incidence of macular edema. Ophthalmology. 1995;102:7-16.
7. Thompson MJ, Ip MS. Diabetic macular edema: a review of past, present, and future therapies. Int Ophthalmol Clin. 2004;44:51-67.
8. Greenstein VC, Shapiro A, Zaidi Q, Hood DC. Psychophysical evidence for post-receptoral sensitivity loss in diabetics. Invest Ophthalmol Vis Sci. 1992;33:2781-90.
9. Parisi V, Uccioli L. Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev. 2001;17:12-8.
10. Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253-262.
11. Bearse MA Jr, Han Y, Schneck ME, Barez S, Jacobsen C, Adams AJ. Local multifocal oscillatory potential abnormalities in diabetes and early diabetic retinopathy. Invest Ophthalmol Vis Sci. 2004;45:3259-65.
12. Caldwell RB, Bartoli M, Behzadian MA, et al. Vascular endothelial growth factor and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev. 2003;19:442-455.
13. Miyamoto K, Hiroshiba N, Tsujikawa A, Ogura Y. In vivo demonstration of increased leukocyte entrapment in retinal microcirculation of diabetic rats. Invest Ophthalmol Vis Sci. 1998;39:2190-2194.
14. Comer GM, Ciulla TA. Pharmacotherapy for diabetic retinopathy. Curr Opin Ophthalmol. 2004;15:508-518.
15. Sheetz MJ, King GL. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA. 2002;288:2579-2588.
16. Fong DS, Aiello LP, Ferris FL 3rd, Klein R. Diabetic retinopathy. Diabetes Care. 2004;27:2540-2553.
17. Kuroki M, Voest EE, Amano S, et al. Reactive oxygen intermediates increase vascular endothelial growth factor expression in vitro and in vivo. J Clin Invest. 1996;98:1667-1675.
18. Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219-1224.
19. McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642-653.
20. Ben-Mahmud BM, Chan WH, Abdulahad RM, et al. Clinical validation of a link between TNF-alpha and the glycosylation enzyme core 2 GlcNAc-T and the relationship of this link to diabetic retinopathy. Diabetologia. 2006;49:2185-2191.
21. Early Treatment Diabetic Retinopathy Study Research Group. Grading diabetic retinopathy from stereoscopic color fundus photographs—an extension of the modified Airlie House classification. ETDRS report number 10. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology. 1991;98(Suppl 5):786-806.
22. Hutchinson A, McIntosh A, Peters J, et al. Effectiveness of screening and monitoring tests for diabetic retinopathy—a systematic review. Diabet Med. 2000;17:495-506.
23. Cavallerano JD, Aiello LP, Cavallerano AA, et al, for the Joslin Vision Network Clinical Team. Nonmydriatic digital imaging alternative for annual retinal examination in persons with previously documented no or mild diabetic retinopathy. Am J Ophthalmol. 2005;140:667-673.
24. Yang CS, Cheng CY, Lee FL, Hsu WM, Liu JH. Quantitative assessment of retinal thickness in diabetic patients with and without clinically significant macular edema using optical coherence tomography. Acta Ophthalmol Scand. 2001;79:266-270.
25. Kim BY, Smith SD, Kaiser PK. Optical coherence tomographic patterns of diabetic macular edema. Am J Ophthalmol. 2006;142:405-412.
26. Kang SW, Park CY, Ham DI. The correlation between fluorescein angiographic and optical coherence tomographic features in clinically significant diabetic macular edema. Am J Ophthalmol. 2004;137:313-322.
27. Ozdek SC, Erdinc MA, Gurelik G, Aydin B, Bahceci U, Hasanreisoglu B. Optical coherence tomographic assessment of diabetic macular edema: comparison with fluorescein angiographic and clinical findings. Ophthalmologica. 2005;219:86-92.
28. Larsson J, Zhu M, Sutter F, Gillies MC. Relation between reduction of foveal thickness and visual acuity in diabetic macular edema treated with intravitreal triamcinolone. Am J Ophthalmol. 2005;139:802-806.
29. Brown DM, Fong D. The correlation of OCT-measured retinal thickness and visual acuity in eyes with diabetic macular edema (DME). Program and abstracts of the 38th Annual Scientific Meeting of the Retina Society; September 15-18, 2005; Coronado, Calif.
30. Kisilevsky M, Hudson C, Flanagan JG, et al. Agreement of the Heidelberg Retina Tomograph II macula edema module with fundus biomicroscopy in diabetic maculopathy. Arch Ophthalmol. 2006;124:337-342.
31. Schatz H, Madeira D, McDonald HR, Johnson RN. Progressive enlargement of laser scars following grid laser photocoagulation for diffuse diabetic macular edema. Arch Ophthalmol. 1991;109:1549-1551.
32. Fong DS, for the Diabetic Retinopathy Clinical Research Network. Results of a randomized trial of focal laser photocoagulation vs. modified grid. Presented at the 2006 American Academy of Ophthalmology Retina Subspecialty Meeting, Retina 2006: Emerging New Concepts; November 11, 2006; Las Vegas, Nev.
33. Friberg TR, Karatza EC. The treatment of macular disease using a micropulsed and continuous wave 810-nm diode laser. Ophthalmology. 1997;104:
2030-2038.
34. Moorman CM, Hamilton AM. Clinical applications of the MicroPulse diode laser. Eye. 1999;13:145-150.
35. McDonald HR, Schatz H. Grid photocoagulation for diffuse macular edema. Retina. 1985;5:65-72.
36. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581-611.
37. Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853-865.
38. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359-371.
39. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:
1306-1309.
40. Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024-1028.
41. Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964-3972.
42. Csaky KG, Baffi JZ, Byrnes GA, et al. Recruitment of marrow-derived endothelial cells to experimental choroidal neovascularization by local expression of vascular endothelial growth factor. Exp Eye Res. 2004;78:1107-1116.
43. Clauss M, Gerlach M, Gerlach H, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. 1990;172:1535-1545.
44. Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood.1996;87:3336-3343.
45. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131-3139.
46. Uhlmann S, Friedrichs U, Eichler W, Hoffmann S, Wiedemann P. Direct measurement of VEGF-induced nitric oxide production by choroidal endothelial cells. Microvasc Res. 2001;62:179-189.
47. Lamoreaux WJ, Fitzgerald ME, Reiner A, Hasty KA, Charles ST. Vascular endothelial growth factor increases release of gelatinase A and decreases release of tissue inhibitor of metalloproteinases by microvascular endothelial cells in vitro. Microvasc Res. 1998;55:29-42.
48. Hiratsuka S, Nakamura K, Iwai S, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289-300.
49. Pepper MS, Ferrara N, Orci L, Montesano R. Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem Biophys Res Commun. 1991;181:902-906.
50. Ng EW, Adamis AP. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can J Ophthalmol. 2005;40:
352-368.
51. Tolentino MJ, Miller JW, Gragoudas ES, et al. Intravitreous injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology. 1996;103:1820-1828.
52. Tolentino MJ, McLeod DS, Taomoto M, Otsuji T, Adamis AP, Lutty GA. Pathologic features of vascular endothelial growth factor-induced retinopathy in the nonhuman primate. Am J Ophthalmol. 2002;133:373-385.
53. Ryan AM, Eppler DB, Hagler KE, et al. Preclinical safety evaluation of rhuMAbVEGF, an antiangiogenic humanized monoclonal antibody. Toxicol Pathol. 1999;27:78-86.
54. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623-628.
55. Fraser HM, Wilson H, Rudge JS, Wiegand SJ. Single injections of vascular endothelial growth factor trap block ovulation in the macaque and produce a prolonged, dose-related suppression of ovarian function. J Clin Endocrinol Metab. 2005;90:1114-1122.
56. Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707-716.
57. Kitamoto Y, Tokunaga H, Tomita K. Vascular endothelial growth factor is an essential molecule for mouse kidney development: glomerulogenesis and nephrogenesis. J Clin Invest. 1997;99:2351-2357.
58. Deodato B, Arsic N, Zentilin L, et al. Recombinant AAV vector encoding human VEGF165 enhances wound healing. Gene Ther. 2002;9:777-785.
59. Nissen NN, Polverini PJ, Koch AE, Volin MV, Gamelli RL, DiPietro LA. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am J Pathol. 1998;152:1445-1452.
60. Arsic N, Zacchigna S, Zentilin L, et al. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther. 2004;10:844-854.
61. LeCouter JR, Moritz DR, Li, B et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299:890-893.
62. Liu MH, Jin H, Floten HS, Ren Z, Yim AP, He GW. Vascular endothelial growth factor-mediated, endothelium-dependent relaxation in human internal mammary artery. Ann Thorac Surg. 2002;73:819-824.
63. Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26:
943-954.
64. Storkebaum E, Lambrechts D, Dewerchin M, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85-92.
65. Ng E, Nishijima K, Robinson GS, Adamis AP, Shima DT. VEGF has both direct and indirect neuroprotective effects in ischemic retina. Invest Ophthalmol Vis Sci. 2006;47:E-Abstract 4829.
66. Kamba T, Tam BY, Hashizume H, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560-576.
67. Marneros AG, Fan J, Yokoyama Y, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167:1451-1459.
68. Blaauwgeers HG, Holtkamp GM, Rutten H, et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol. 1999;155:421-428.
69. Gordon MS, Cunningham D. Managing patients treated with bevacizumab combination therapy. Oncology. 2005;69(Suppl 3):25-33.
70. Cannistra SA, Matulonis U, Penson R, et al. Bevacizumab in patients with advanced platinum-resistant ovarian cancer. J Clin Oncol. 2006;24(Suppl 18):Abstract 5006.
71. Skillings JR, Johnson DH, Miller K, et al. Arterial thromboembolic events (ATEs) in a pooled analysis of 5 randomized, controlled trials (RCTs) of bevacizumab (BV) with chemotherapy. J Clin Oncol. 2005;23(Suppl 16S):Abstract 3019.
72. Avastin® Prescribing Information; Genentech, Inc. Available at: http://www.gene.com/gene/products/information/oncology/avastin/insert.jsp. Accessed October 8, 2006.
73. Fung AD, Rosenfeld PJ, Reichel E. The International Intravitreal Bevacizumab Safety Survey: using the Internet to assess drug safety worldwide. Br J Ophthalmol. 2006;90:1344-1349.
74. van Wijngaarden P, Coster DJ, Williams KA. Inhibitors of ocular neovascularization: promises and potential problems. JAMA. 2005;293:1509-1513.
75. Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845-848.
76. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843-845.
77. Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. 1995;113:1538-1544.
78. Famiglietti EV, Stopa EG, McGookin ED, Song P, LeBlanc V, Streeten BW. Immunocytochemical localization of vascular endothelial growth factor in neurons and glial cells of human retina. Brain Res. 2003;969:195-204.
79. Shima DT, Adamis AP, Ferrara N, et al. Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol Med. 1995;1:182-193.
80. Senger DR, Connolly DT, Van de Water L, Feder J, Dvorak HF. Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res. 1990;50:1774-1778.
81. Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108:2369-2379.
82. Antonetti D, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. J Biol Chem. 1999;274:23463-23467.
83. Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Ultrastructural studies define soluble macromolecular, particulate, and cellular transendothelial cell pathways in venules, lymphatic vessels, and tumor-associated microvessels in man and animals. Microsc Res Tech. 2002;57:289-326.
84. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497-504.
85. Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480-1487.
86. Funatsu H, Yamashita H, Sakata K, et al. Vitreous levels of vascular endothelial growth factor and intercellular adhesion molecule 1 are related to diabetic macular edema. Ophthalmology. 2005;112:806-816.
87. Qaum T, Xu Q, Joussen AM, et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408-2413.
88. Joussen A, Murata T, Tsujikawa A, Kirchhof B, Bursell S-E, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147-152.
89. Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501-509.
90. Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438-440.
91. Joussen AM, Poulaki V, Mitsiades N, et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. 2003;17:76–78.
92. Ishida S, Usui T, Yamashiro K, et al. VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci. 2003;44:2155-2162.
93. Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003;198:483-489.
94. Gragoudas ES, Adamis AP, Cunningham ET, Jr., Feinsod M, Guyer DR, for the VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group. Pegaptanib for neovascular age-related macular degeneration. N Engl JMed. 2004;351:2805-2816.
95. Chakravarthy U, Adamis AP, Cunningham ET, Jr., et al, for the VEGF Inhibition Study in Ocular Neovascularization (V.I.S.I.O.N.) Clinical Trial Group. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology. 2006;113:
1508.e1-25.
96. D'Amico DJ, Patel M, Adamis AP, Cunningham ET, Jr., Guyer DR, Katz B, for the VEGF Inhibition Study in Ocular Neovascularization (V.I.S.I.O.N.) Clinical Trial Group. Pegaptanib sodium for neovascular age-related macular degeneration: two-year safety results of the two prospective, multicenter, controlled clinical trials. Ophthalmology. 2006;113:1001.e1-6.
97. Suner IJ. Safety of pegaptanib sodium in age-related macular degeneration
(AMD): 3-year results of the VISION trial. Poster presented at the 2006 annual meeting of the American Academy of Ophthalmology; November 13, 2006; Las Vegas, Nev.
98. Cunningham ET, Jr., Adamis AP, Altaweel M, et al, for the Macugen Diabetic Retinopathy Study Group. A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology. 2005;112:1747-1757.
99. Adamis AP, Altaweel M, Bressler NM, et al. Macugen Diabetic Retinopathy Study Group. Changes in retinal neovascularization after pegaptanib (Macugen) therapy in diabetic individuals. Ophthalmology. 2006;113:23-28.
100. Gonzalez V. Selective VEGF inhibition: effectiveness in modifying the progression of proliferative diabetic retinopathy. Poster presented at the 2006 annual meeting of the American Academy of Ophthalmology; November 12, 2006; Las Vegas, Nev.
101. Ferrara N, Damico L, Shams N, Lowman H, Kim R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina. 2006;26:859-870.
102. Brown DM, Kaiser PK, Michels M, et al, for the ANCHOR Study Group. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432-1444.
103. Rosenfeld PJ, Brown DM, Heier JS, et al, for the MARINA Study Group. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419-1431.
104. Nguyen QD, Tatlipinar S, Shah SM, et al. Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol. 2006 Aug 2; [Epub ahead of print].
105. Chun DW, Heier JS, Topping TM, Duker JS, Bankert JM. A pilot study of multiple intravitreal injections of ranibizumab in patients with center-involving clinically significant diabetic macular edema. Ophthalmology. 2006;113:
1706-1712.
106. Arevalo JF, Fromow-Guerra JJ, Sanchez JG, et al. Primary intravitreal bevacizumab (Avastin) for diabetic macular edema: results of a multicenter collaborative study group. Presented at the 2006 annual meeting of the American Academy of Ophthalmology; November 12, 2006; Las Vegas, Nev.
107. Oshima Y, Sakaguchi H, Gomi F, Tano Y Regression of iris neovascularization after intravitreal injection of bevacizumab in patients with proliferative diabetic retinopathy. Am J Ophthalmol. 2006;142:155-158.
108. Mason JO 3rd, Nixon PA, White MF. Intravitreal injection of bevacizumab (Avastin) as adjunctive treatment of proliferative diabetic retinopathy. Am J Ophthalmol. 2006;142:685-688.
109. Avery RL, Pearlman J, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ, Wendel R, Patel A. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmology. 2006;113:
1695.e1-15.
110. Xia P, Aiello LP, Ishii H, et al. Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest. 1996;98:2018-2026.
111. Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473-1480.
112. Bursell SE, Takagi C, Clermont AC, et al. Specific retinal diacylglycerol and protein kinase C beta isoform modulation mimics abnormal retinal hemodynamics in diabetic rats. Invest Ophthalmol Vis Sci. 1997;38:2711-2720.
113. Xu X, Zhu Q, Xia X, Zhang S, Gu Q, Luo D. Blood-retinal barrier breakdown induced by activation of protein kinase C via vascular endothelial growth factor in streptozotocin-induced diabetic rats. Curr Eye Res. 2004;28:251-256.
114. PKC-DRS Group. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C Beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes. 2005;54:2188-2197.
115. Strom C, Sander B, Klemp K, Aiello LP, Lund-Andersen H, Larsen M. Effect of ruboxistaurin on blood-retinal barrier permeability in relation to severity of leakage in diabetic macular edema. Invest Ophthalmol Vis Sci. 2005;46: 3855-3858.
116. Aiello LP, Clermont A, Arora V, Davis MD, Sheetz MJ, Bursell SE. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Invest Ophthalmol Vis Sci. 2006;47:86-92.
117. PKC-DRS2 Group. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006 Sept 19; [article in press] [Epub ahead of print].
118. OSN SuperSite. Retina/Vitreous. Lilly weighing ruboxistaurin options as FDA requests new trial. Available at: http://www.osnsupersite.com/view.asp?rID=
18630. Accessed November 9, 2006.
119. Eli Lilly and Company. Lilly announces FDA requirement of additional clinical trial before ruboxistaurin could be approved for treatment of diabetic retinopathy. Available at: http://newsroom.lilly.com/ReleaseDetail.cfm?
ReleaseID=212654. Accessed on November 9, 2006.
120. Penfold PL, Wen L, Madigan MC, King NJ, Provis JM. Modulation of permeability and adhesion molecule expression by human choroidal endothelial cells. Invest Ophthalmol Vis Sci. 2002;43:3125-3130.
121. Matsuda S, Gomi F, Oshima Y, Tohyama M, Tano Y. Vascular endothelial growth factor reduced and connective tissue growth factor induced by triamcinolone in ARPE19 cells under oxidative stress. Invest Ophthalmol Vis Sci. 2005;46:1062-1068.
122. Danis RP, Bingaman DP, Yang Y, Ladd B. Inhibition of preretinal and optic nerve head neovascularization in pigs by intravitreal triamcinolone acetonide. Ophthalmology. 1996;103:2099-2104.
123. Jonas JB. Intravitreal triamcinolone acetonide: a change in a paradigm. Ophthalmic Res. 2006;38:218-245.
124. Kaiser PK. Verteporfin therapy in combination with triamcinolone: published studies investigating a potential synergistic effect. Curr Med Res Opin. 2005;21:705-713.
125. Jonas JB, Kamppeter BA, Harder B, Vossmerbaeumer U, Sauder G, Spandau UH. Intravitreal triamcinolone acetonide for diabetic macular edema: a prospective, randomized study. J Ocul Pharmacol Ther. 2006;22:200-207.
126. Gillies MC, Sutter FK, Simpson JM, Larsson J, Ali H, Zhu M. Intravitreal triamcinolone for refractory diabetic macular edema: two-year results of a double-masked, placebo-controlled, randomized clinical trial. Ophthalmology. 2006;113:1533-1538.
127. Adis International Limited. Fluocinolone acetonide ophthalmic — Bausch & Lomb: fluocinolone acetonide Envision TD implant. Drugs R D. 2005;6:116-119.
128. Kuppermann BD, Blumenkranz MS, Haller JA, Williams GA; the Posurdex Study Group. An intravitreous dexamethasone biodegradible drug delivery system for the treatment of persistent diabetic macular edema. Invest Ophthalmol Vis Sci. 2003;44:E-Abstract 4289.
129. Fong DS, Aiello LP, Ferris FL, 3rd, Klein R. Diabetic retinopathy. Diabetes Care. 2004; 27:2540-2553.
130. Siddiqui MA, Scott LJ. Infliximab: a review of its use in Crohn's disease and rheumatoid arthritis. Drugs. 2005;65:2179-2208.
131. Ben-Mahmud BM, Mann GE, Datti A, Orlacchio A, Kohner EM, Chibber R. Tumor necrosis factor-alpha in diabetic plasma increases the activity of core 2 GlcNAc-T and adherence of human leukocytes to retinal endothelial cells: significance of core 2 GlcNAc-T in diabetic retinopathy. Diabetes. 2004;53:2968-2976.
132. Hangai M, He S, Hoffmann S, Lim JI, Ryan SJ, Hinton DR. Sequential induction of angiogenic growth factors by TNF-alpha in choroidal endothelial cells. J Neuroimmunol. 2006;171:45-56.
133. Sfikakis PP, Markomichelakis N, Theodossiadis GP, Grigoropoulos V, Katsilambros N, Theodossiadis PG. Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care. 2005;28:445-447.
134. Markomichelakis NN, Theodossiadis PG, Sfikakis PP. Regression of neovascular age-related macular degeneration following infliximab therapy. Am J Ophthalmol. 2005;139:537-540.
| Victor Gonzalez, MD, is the principal of the Valley Retina Institute in McAllen, Texas. He can be reached at maculadoc@aol.com. Dr. Gonzalez is a consultant for Pfizer, Inc, and has received a research grant from the company. |