







peer reviewed
Advances in the Management of Retinoblastoma
CAROL L. SHIELDS, MD • JERRY
A. SHIELDS, MD
Retinoblastoma represents approximately 4% of all pediatric malignancies and is the most common intraocular malignancy in children.1,2 It is estimated that 300 new cases of retinoblastoma are diagnosed in the United States each year and 5000 cases worldwide. Over 95% of children with retinoblastoma in the United States and other medically developed nations survive their malignancy, whereas about 50% survive worldwide.
Retinoblastoma can be classified in 3 different ways: familial or sporadic, bilateral or unilateral, and heritable or nonheritable. About two-thirds of all cases are unilateral and one-third are bilateral. Genetically, it is simpler to discuss retinoblastoma with the latter classification of heritable (germline mutation) or nonheritable (somatic mutation). Bilateral and familial retinoblastoma are caused by a germline mutation and are thus heritable. Unilateral sporadic retinoblastoma is usually not heritable, but approximately 15% of patients will show a germline mutation. Genetic testing using DNA analysis of the patient's tumor and peripheral blood can help identify those patients with germline mutation.
FIRST, REMEMBER THE LIFE THREATENING ASPECTS OF RETINOBLASTOMA
Children with retinoblastoma are at risk for 3 important, life threatening problems including metastasis from retinoblastoma, intracranial neuroblastic malignancy (trilateral retinoblastoma), and second primary tumors.
Retinoblastoma metastasis typically develops within 1 year of the diagnosis of the intraocular tumor. Those at greatest risk for metastasis show features of retinoblastoma invasion beyond the lamina cribrosa in the optic nerve, in the choroid, sclera, orbit, or anterior chamber.3 Patients with invasive retinoblastoma should be treated with chemotherapy for 4 to 6 months to prevent metastases.
Patient with hereditary retinoblastoma are at risk for neuroblastic intracranial malignancy in the first 5 years of life, most often manifesting as pinealoblastoma or other parasellar tumors.4 This has been termed "trilateral" retinoblastoma and is found in approximately 3% of all children with retinoblastoma. Unfortunately, pinealoblastoma is usually fatal with few survivors. Systemic chemoreduction for retinoblastoma may prevent trilateral retinoblastoma.
Genetically-related second cancers can occur in survivors of bilateral or heritable retinoblastoma.5,6 Patients with hereditary retinoblastoma have approximately a 5% chance of developing a second cancer during the first 10 years of follow-up, 18% during the first 20 years, and 26% within 30 years.5 Second cancers most often include osteogenic sarcoma, spindle cell sarcoma, chondrosarcoma, rhabdomyosarcoma, neuroblastoma, glioma, leukemia, sebaceous cell carcinoma, squamous cell carcinoma, and malignant melanoma.
WHAT ARE THE OPHTHALMOSCOPIC FEATURES OF RETINOBLASTOMA?
The clinical manifestations of retinoblastoma vary with the stage of the disease.1,2 A small retinoblastoma <2 mm in diameter appears transparent or slightly translucent in the sensory retina.1,2 Larger tumors stimulate dilated retinal blood vessels feeding the tumor, foci of intrinsic calcification and can produce subretinal fluid (exophytic pattern), subretinal seeding, and vitreous seeding (endophytic pattern). Retinoblastoma of any size can produce leukocoria, but this is most often seen with large tumors.
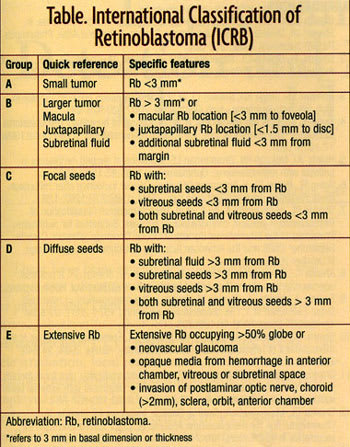
HOW DO WE CLASSIFY RETINOBLASTOMA?
Several classifications of retinoblastoma have been developed including the Reese Ellsworth classification and the more recent International Classification of Retinoblastoma (ICRB) (Table). The ICRB is useful in that it can assist in prediction of chemoreduction success.7
KEEP IN MIND THE DIFFERENTIAL DIAGNOSIS
A number of ocular disorders in infants and children can resemble retinoblastoma. The most common pseudoretinoblastomas included persistent hyperplastic primary vitreous, Coats' disease, and ocular toxocariasis. It is important that the diagnosis of retinoblastoma be excluded without question before treatment of a pseudoretinoblastoma. Vitrectomy should be withheld until the diagnosis of retinoblastoma is reliably excluded. Consultation with ocular oncologists experienced with retinoblastoma could be helpful in confirming the suspected clinical diagnosis and excluding retinoblastoma.
WHAT ARE THE CHOICES FOR MANAGEMENT OF RETINOBLASTOMA?
Management of retinoblastoma is tailored to each individual case and based on the overall situation, including threat of metastatic disease, risks for second cancers, systemic status, laterality of the disease, size and location of the tumor(s), and estimated visual prognosis. The currently available treatment methods include intravenous chemoreduction (carboplatin, etoposide, and vincristine) subconjunctival carboplatin, thermotherapy, cryotherapy, laser photocoagulation, plaque radiotherapy, external beam radiotherapy, and enucleation,8,9 For unilateral retinoblastoma, enucleation is performed in 75% of cases and conservative treatment (nonenucleation methods) is possible in 25%. The reason for the high rate of enucleation is that unilateral retinoblastoma is typically detected when the tumor is advanced. For those with less advanced disease, chemoreduction plus focal consolidation of each tumor with thermotherapy or cryotherapy, or the use of plaque radiotherapy or other methods are beneficial. For bilateral retinoblastoma, chemoreduction plus thermotherapy or cryotherapy is necessary in most cases and about 60% of patients eventuate in loss of one eye.10-12 Enucleation of both eyes is only necessary in 1% of cases. Based on the ICRB, chemoreduction success is achieved in 100% of group A, 93% of group B, 90% of group C, and 47% of group D eyes (Figure 1).7 Group D eyes often require the addition of subconjunctival carboplatin as a local boost in chemotherapy.13
CAN TUMORS RECUR AFTER CHEMOREDUCTION?
The main problem with chemoreduction is recurrence of vitreous or subretinal seeds, usually remote from the main tumors.10-12 These seeds generally respond to initial chemoreduction, but later they can recur. and require treatment with cryotherapy, thermotherapy, plaque radiotherapy, external beam radiotherapy, or enucleation. It should also be realized that 24% of patients develop new retinoblastomas during or after chemoreduction, generally in those who present as infants and with family history of retinoblastoma.
CAN PLAQUE RADIOTHERAPY BE USED FOR RETINOBLASTOMA?
|
|
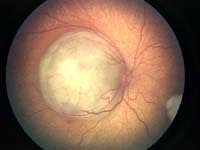
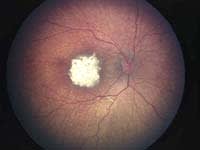
| Figure 1. Regression of macular retinoblastoma
following chemoreduction and focal foveal-sparing thermotherapy. Top: Before treatment. Bottom: After treatment, the tumor has remained regressed without recurrence at 5 years. |
Plaque radiotherapy is a method of brachytherapy in which a radioactive implant is placed on the sclera over the base of a retinoblastoma to irradiate the tumor transclerally. It is limited to tumors <16 mm in base and 8 mm in thickness and complete treatment can be achieved in approximately 4 days. Plaque radiotherapy provides long-term tumor control in 90% of eyes when used as a primary treatment.14 In those eyes that need plaque radiotherapy for recurrence after chemoreduction, complete control of the tumor is achieved in 96% of cases. All eyes treated with plaque radiotherapy should be monitored for radiation maculopathy and papillopathy.
WHEN IS ENUCLEATION USED?
Enucleation is an important method for managing retinoblastoma. Enucleation is employed for advanced disease with no hope for useful vision in the affected eye or if there is a concern for invasion of the tumor into the optic nerve, choroid, or orbit.
The technique of enucleation is to gently remove the eye intact without seeding the malignancy into the orbit. After the globe is removed, fresh tissue is harvested on a separate tray in the operating room for DNA analysis, using a specific technique. The surgeon harvesting fresh tissue must remove the used gloves and reglove after this step to avoid the risk of tumor contamination into the child's orbit. If spillage onto the surgeons gown occurs, then complete regowning and regloving is necessary. Following enucleation, an orbital implant is placed to provide a more natural cosmetic appearance of the patient's artificial eye and to enable motility of the prosthesis.15 There are several available orbital implants including polymethylmethacrylate sphere, coralline hydroxyapatite, polyethylene, and others.
In summary, retinoblastoma continues to be a challenge both diagnostically and therapeutically. It is important to first clearly establish the correct diagnosis before embarking on therapy. Many factors enter into the decision regarding management such as patient age, tumor laterality, size, location, extent, and anticipated visual prognosis.
Chemoreduction is an important conservative method for tumor control. Enucleation still proves to be useful for advanced tumor.
REFERENCES
1. Shields JA, Shields CL. Management and prognosis of retinoblastoma. In Shields JA, Shields CL., eds. Intraocular Tumors. A Text and Atlas. Philadelphia, Pa: WB Saunders; 1992; 377-392.
2. Shields JA, Shields CL. Retinoblastoma. In Shields JA, Shields CL, eds. Atlas of Intraocular Tumors. Philadelphia, Pa: Lipincott Williams Wilkins; 1999:207-232.
3. Honavar SG, Singh AD, Shields CL,et al. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120:923-931.
4. Kivela T. Trilateral retinoblastoma: A meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol.1999; 17:1829-1837.
5. Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in patients with retinoblastoma. Ophthalmology 1988;95:1583-1587.
6. Wong FL, Boice JD Jr, Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262-1267.
7. Shields CL, Mashayekhi A, Au A, et al. The International Classification of Retinoblastoma predicts chemoreduction success. Submitted for publication. Presented at the International Society of Ocular Oncology; Whistler, Canada; September 2005 and the American Academy of Ophthalmology; Chicago, Ill;October 2005.
8. Shields CL, Mashayekhi A, Demirci H, Meadows AT, Shields JA. A practical approach to management of retinoblastoma. Arch Ophthalmol. 2004. In press.
9. Epstein J, Shields CL, Shields JA. Trends in the management of retinoblastoma; Evaluation of 1,196 consecutive eyes during 1974-2001. J Pediatr Ophthalmol Strab. 2003;40:196-203.
10. Shields CL, Meadows AT, Leahey AM, Shields JA. Continuing challenges in the management of retinoblastoma with chemotherapy. Retina. 2004; 24:849-862.
11. Shields CL, Honavar SG Meadows AT, et al. Chemoreduction plus focal therapy for retinoblastoma: Factors predictive of need for treatment with external beam radiotherapy or enucleation. Am J Ophthalmol. 2002;133:657-664.
12. Shields CL, Mashayekhi A, Cater J, Shelil A, Meadows AT, Shields JA. Chemoreduction for retinoblastoma Analysis of tumor control and risks for recurrence in 457 tumors. Am J Ophthalmol. 2004;138:329-337.
13. Abramson DH, Frank CM, Dunkel IJ. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. Ophthalmology. 1999;106:1947-1950.
14. Shields CL, Shields JA, Cater J, Othmane I, Singh AD, Micaily B. Plaque radiotherapy for retinoblastoma: long term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116-2121.
15. Shields CL, Shields JA, DePotter P, Singh AD. Lack of complications of the hydroxyapatite orbital implant in 250 consecutive cases. Trans Am Ophthalmol Soc. 1993;91:177-189.
Jerry Shields, MD, is director, oncology services at Wills Eye Hospital and professor of ophthalmology at Thomas Jefferson University. Carol Shields, MD, is co-director, oncology services at Wills Eye Hospital and professor of ophthalmology at Thomas Jefferson University. Support provided by the Macula Foundation, New York, NY and the Eye Tumor Research Foundation, Philadelphia, PA. Neither author has any financial interest in the information contained in this article.